- 英
- Angelman syndrome AS
- 同
- Angelman症候群、アンゲルマン症候群、愉快な人形症候群 happy puppet症候群 happy puppet syndrome
- 関
- プラダー・ウィリー症候群
概念・起因
- 約60%に15q11-13の微小欠失を検出し、欠失は母親由来。一部に父親由来の片親性ダイソミー。Prader-Willi症候群とは病因遺伝子が異なるが、欠失は近傍に検出され、かつ由来が異なる。
症状
- 笑い発作、重度の知的障害、特異な顔貌(下顎突出など)、色白、外斜視、操り人形様の欠調性歩行。
ASとPWSの比較 (PED.215改変)
| PWSに特徴的 | 共通症状 | ASに特徴的 | ||
| 高度肥満 | 11q11-a12の欠失 | 操り人形様の失調性歩行 | ||
| 異常な食欲 | 精神発達遅滞 | より高度 | 容易に誘発される発作的な笑い | |
| 乳幼児期の筋緊張低下 | 痛覚鈍麻 | 手足の振戦 | ||
| 小さな手足と先細りの指 | より高度 | 色素脱出 | 小頭症 | |
| 性腺機能低下 | 痙攣 | 高頻度 | 大きな突出した下顎 | |
| より高度 | 肥満 | 流涎を伴う舌挺出 | ||
PrepTutorEJDIC
- Anglo-Saxon
Wikipedia preview
出典(authority):フリー百科事典『ウィキペディア(Wikipedia)』「2013/05/13 13:48:41」(JST)
wiki ja
UpToDate Contents
全文を閲覧するには購読必要です。 To read the full text you will need to subscribe.
- 1. 微細欠失症候群(12番~22番染色体) microdeletion syndromes chromosomes 12 to 22
- 2. ゲノム疾患:概要 genomic disorders an overview
- 3. 自閉症スペクトラム障害:診断 autism spectrum disorder diagnosis
- 4. 幼児および小児における痙攣発作の臨床検査診断 clinical and laboratory diagnosis of seizures in infants and children
- 5. プラダーウィリ症候群の疫学および遺伝学 epidemiology and genetics of prader willi syndrome
Japanese Journal
- アンジェルマン症候群の中枢神経機能障害
- 齋藤 伸治
- 日本小児科学会雑誌 116(1), 40-45, 2012-01-01
- NAID 10030289376
- 小児の脳機能発達とエピジェネティクス : アンジェルマン症候群をモデルとして
- 齋藤 伸治
- 日本周産期・新生児医学会雑誌 = Journal of Japan Society of Perinatal and Neonatal Medicine 47(4), 755-757, 2011-12-20
- NAID 10030287598
- 連日生じるミオクロニー重積に静注用フェノバルビタールが著効したアンジェルマン症候群の1成人例
- 永井 達哉,齊藤 聖,高木 俊輔,坂田 増弘,渡辺 雅子,渡辺 裕貴
- てんかん研究 = Journal of the Japan Epilepsy Society 29(1), 44-51, 2011-06-30
- … 本症例は非定型欠神発作、頻繁な微笑なども認められ、染色体検査にて15q11-13の欠失が確認され、アンジェルマン症候群の診断が確定した。 … アンジェルマン症候群ではミオクロニー発作や非定型欠神発作が多く、重積になりやすい。 … これらの発作に精神遅滞を伴う場合、アンジェルマン症候群を鑑別に入れることが重要であり、フェノバルビタールが著効する場合がある。 …
- NAID 10029651189
- 障害児の楽器の認知過程に関する一考察--アンジェルマン症候群児への音楽療法を通して
- 馬場 悦子
- 純真紀要 (51), 45-54, 2010-12-00
- NAID 40018871684
Related Links
- アンジェルマン症候群(AS)は,重度の運動発達遅滞 や精神遅滞 ,重度の言語 障害,失調性歩行や四肢の振戦 ,独特の行動を特徴とする15番染色体上の一部の遺伝子の欠失症です。比較的まだ新しい病気で情報が少ないですが ...
- GRJ top > 遺伝子疾患情報リスト アンジェルマン症候群 (Angelman Syndrome) Gene Review著者: Aditi I Dagli, MD,Charles A Williams, MD 日本語訳者: 窪田美穂(ボランティア翻訳者),石川亜貴(札幌医科大学医学部遺伝医学) ...
- はじめに 1965年, 英国の医師ヘリー・アンジェルマン(Harry Angelman)博士が,現在アンジェルマン症候群(AS)として知られるところとなった特徴的症状を示す3名の子供に関する記述を初めて行いました 1 .博士は,どの子供もこわばった ...
Related Pictures




★リンクテーブル★
| リンク元 | 「プラダー・ウィリー症候群」「15番染色体」「AS」「片親性ダイソミー」「happy puppet syndrome」 |
| 関連記事 | 「症候群」「群」「症候」 |
「プラダー・ウィリー症候群」
- 英
- Prader-Willi syndrome, PWS
- 同
- Prader-Willi症候群、プラダー-ウィリー症候群、プラダー・ウィリ症候群 プラダーウィリー症候群、プラダー・ウィリィ症候群、プラダー-ラープハルト-ウィリー症候群 Prader-Labhart-Willi syndrome PLWS
- 潜伏精巣・低身長症・肥満・軽度精神発達遅滞症候群 cryptorchidism-dwarfism-obesity-subnormal mentality syndrome、筋緊張低下・知能低下・性腺発育不全・肥満症候群 hypotonia-hypomentia-hypogonadism-obesity syndrome HHHO症候群 HHHO syndrome H3O syndrome
- 関
- アンジェルマン症候群
概念
- 15番染色体長腕の動原体近傍の染色体の腕内欠失による先天性疾患。
- 父親に由来する異常染色体を引き継いだ場合に発症する(ゲノムインプリンティング)。 ← そうでない場合もある(see 病因)
病因
- 1. 70%:15番染色体長腕q11-13の父親由来染色体部分欠失
- 2. 20%:母性片親性ダイソミー
症状
| http://schoolworkhelper.net/wp-content/uploads/2011/06/Prader-Willi-Syndrome-2.gif |
|
{kind=link}
ASとPWSの比較 (PED.215改変)
| PWSに特徴的 | 共通症状 | ASに特徴的 | ||
| 高度肥満 | 11q11-a12の欠失 | 操り人形様の失調性歩行 | ||
| 異常な食欲 | 精神発達遅滞 | より高度 | 容易に誘発される発作的な笑い | |
| 乳幼児期の筋緊張低下 | 痛覚鈍麻 | 手足の振戦 | ||
| 小さな手足と先細りの指 | より高度 | 色素脱出 | 小頭症 | |
| 性腺機能低下 | 痙攣 | 高頻度 | 大きな突出した下顎 | |
| より高度 | 肥満 | 流涎を伴う舌挺出 | ||
参考
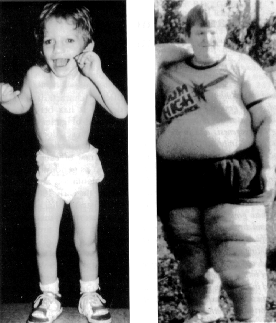
- 1. 写真
{kind=link}
- 2.
「15番染色体」
- 英
- chromosome 15 chr 15
- 同
- 第15番染色体、第15染色体
- 急性前骨髄球性白血病 APLでは染色体異常が見られる。t(15;17)(PML/RARα)
- アンジェルマン症候群、プラダー・ウィリー症候群:15q11-13の微小欠失
「AS」
- アンチセンス antisense
- 大動脈弁狭窄症 大動脈弁狭窄 aortic stenosis
- アンジェルマン症候群 Angelman syndrome
- アルギニノコハク酸合成酵素 argininosuccinate synthetase
- アルポート症候群 Alport syndrome
- 心房静止 atrial standstill
- エトキシスクレロール Aethoxysklerol = ポリドカノール
「片親性ダイソミー」
臨床関連
- プラダー・ウィリー症候群:一部
- アンジェルマン症候群:一部
「happy puppet syndrome」
「症候群」
- 英
- syndrome, symptom-complex
- 同
- 症状群
- 関
- [[]]
- 成因や病理学的所見からではなく、複数の症候の組み合わせによって診断される診断名あるいは疾患。
内分泌
先天的代謝異常
- レッシュ・ナイハン症候群 レッシュ-ナイハン症候群 Lesch-Nyhan症候群:HGPRTの欠損により尿酸が蓄積するX染色体連鎖劣性遺伝疾患であり、痙性麻痺、不随意運動、知的障害、自傷行為が見られる。(QB.D-233)
高プロラクチン血症
- 分娩後の視床下部障害によるプロラクチン分泌抑制因子の分泌抑制のため、高プロラクチン血症を呈する。
- 分娩に関係なくプロラクチン分泌抑制因子の分泌抑制をきたし、高プロラクチン血症を呈する。
性腺機能低下
- 嗅覚の低下・脱出、低ゴナドトロピン性性腺機能低下症
- Bardet-Biedl症候群 バルデー・ビードル症候群 バルデ-ビードル症候群
- Laurence-Moon-Bardet-Biedl症候群 ローレンス-ムーン-バルデ-ビードル症候群
- Laurence-Moon-Biedl症候群 ローレンス・ムーン・ビードル症候群
- 肥満、網膜色素変性症、知能低下、低ゴナドトロピン性性器発育不全、多指症、低身長
性早熟
- 思春期早発症、多発性線維性骨異形成症、皮膚色素沈着
- Sheehan症候群 シーハン症候群:汎下垂体機能低下症。
- 女性型の肥満、性器の発育障害の2主徴を示し、視床下部に器質的障害をもつ疾患群。
脳神経外科・神経内科
- Wallenberg症候群 ワレンベルグ症候群:椎骨動脈、後下小脳動脈の血栓塞栓症などで生じる。頚部より下位で温度覚の障害が健側に出現するのに対し、頚部より上位では障害側に温度覚の障害が出現する。
「群」
「症候」
- 英
- symptom and sign
- 関
- 症状, 徴候 兆候