| Inborn error of metabolism |
| Classification and external resources |
| ICD-10 |
E70-E90 |
| ICD-9-CM |
270-279 |
| MedlinePlus |
002438 |
| eMedicine |
emerg/768 article/804757 |
| MeSH |
D008661 |
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of metabolism. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are now often referred to as congenital metabolic diseases or inherited metabolic diseases.
The term inborn error of metabolism was coined by a British physician, Archibald Garrod (1857–1936), in the early 20th century (1908). He is known for work that prefigured the "one gene-one enzyme" hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism was published in 1923.[1]
Contents
- 1 Classification
- 2 Signs and symptoms
- 3 Diagnosis
- 4 Treatment
- 5 Epidemiology
- 6 References
- 7 External links
Classification
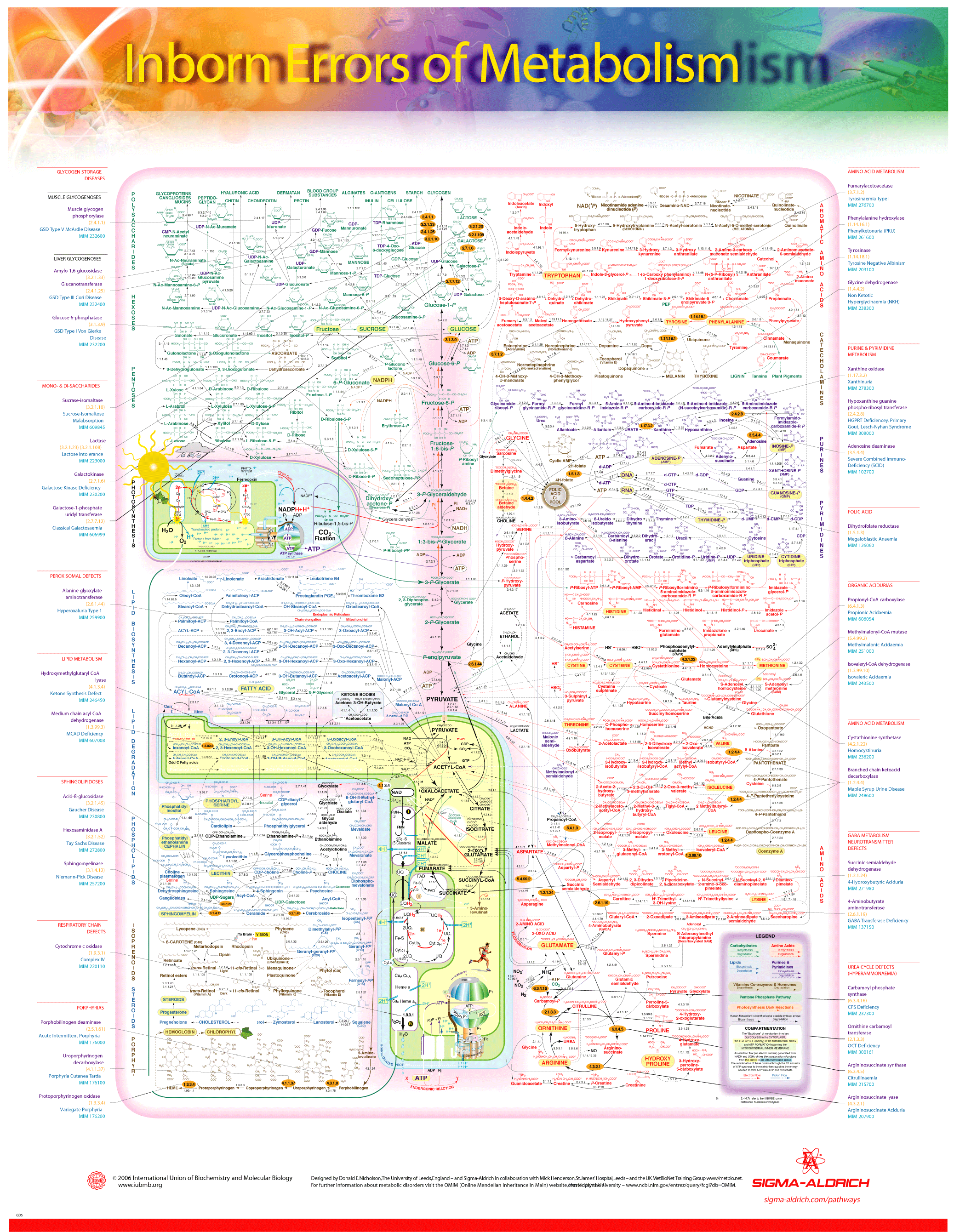
Traditionally the inherited metabolic diseases were classified as disorders of carbohydrate metabolism, amino acid metabolism, organic acid metabolism, or lysosomal storage diseases. In recent decades, hundreds of new inherited disorders of metabolism have been discovered and the categories have proliferated. Following are some of the major classes of congenital metabolic diseases, with prominent examples of each class. Many others do not fall into these categories.
- Disorders of carbohydrate metabolism
- E.g., glycogen storage disease
- Disorders of amino acid metabolism
- E.g., phenylketonuria, maple syrup urine disease, glutaric acidemia type 1
- Urea Cycle Disorder or Urea Cycle Defects
- E.g., Carbamoyl phosphate synthetase I deficiency
- Disorders of organic acid metabolism (organic acidurias)
- E.g., alcaptonuria, 2-hydroxyglutaric acidurias
- Disorders of fatty acid oxidation and mitochondrial metabolism
- E.g., Medium-chain acyl-coenzyme A dehydrogenase deficiency (often shortened to MCADD.)
- Disorders of porphyrin metabolism
- E.g., acute intermittent porphyria
- Disorders of purine or pyrimidine metabolism
- E.g., Lesch-Nyhan syndrome
- Disorders of steroid metabolism
- E.g., lipoid congenital adrenal hyperplasia, congenital adrenal hyperplasia
- Disorders of mitochondrial function
- E.g., Kearns-Sayre syndrome
- Disorders of peroxisomal function
- Lysosomal storage disorders
- E.g., Gaucher's disease
- E.g., Niemann Pick disease
Signs and symptoms
Because of the enormous number of these diseases and wide range of systems affected, nearly every "presenting complaint" to a doctor may have a congenital metabolic disease as a possible cause, especially in childhood. The following are examples of potential manifestations affecting each of the major organ systems: many manifestations may develop
- Growth failure, failure to thrive, weight loss
- Ambiguous genitalia, delayed puberty, precocious puberty
- Developmental delay, seizures, dementia, encephalopathy, stroke
- Deafness, blindness, pain agnosia
- Skin rash, abnormal pigmentation, lack of pigmentation, excessive hair growth, lumps and bumps
- Dental abnormalities
- Immunodeficiency, low platelet count, low red blood cell count, enlarged spleen, enlarged lymph nodes
- Many forms of cancer
- Recurrent vomiting, diarrhea, abdominal pain
- Excessive urination, kidney failure, dehydration, edema
- Low blood pressure, heart failure, enlarged heart, hypertension, myocardial infarction
- Liver enlargement, jaundice, liver failure
- Unusual facial features, congenital malformations
- Excessive breathing (hyperventilation), respiratory failure
- Abnormal behavior, depression, psychosis
- Joint pain, muscle weakness, cramps
- Hypothyroidism, adrenal insufficiency, hypogonadism, diabetes mellitus
Diagnosis
Dozens of congenital metabolic diseases are now detectable by newborn screening tests, especially the expanded testing using mass spectrometry. This is an increasingly common way for the diagnosis to be made and sometimes results in earlier treatment and a better outcome. There is a revolutionary GC/MS based technology with an integrated analytics system, which has now made it possible to test a newborn for over 100 mmgenetic metabolic disorders.
Because of the multiplicity of conditions, many different diagnostic tests are used for screening. An abnormal result is often followed by a subsequent "definitive test" to confirm the suspected diagnosis.
Common screening tests used in the last sixty years:
- Ferric chloride test (turned colors in reaction to various abnormal metabolites in urine)
- Ninhydrin paper chromatography (detected abnormal amino acid patterns)
- Guthrie bacterial inhibition assay (detected a few amino acids in excessive amounts in blood) The dried blood spot can be used for multianalyte testing using Tandem Mass Spectrometry (MS/MS). This given an indication for a disorder. The same has to be further confirmed by enzyme assays, IEX-Ninhydrin, GC/MS or DNA Testing.
- Quantitative measurement of amino acids in plasma and urine
- IEX-Ninhydrin post column derivitization liquid ion-exchange chromatography (detected abnormal amino acid patterns and quantitative analysis)
- Urine organic acid analysis by Gas chromatography-mass spectrometry
- Plasma acylcarnitines analysis by mass spectrometry
- Urine purines and pyrimidines analysis by Gas chromatography-mass spectrometry
Specific diagnostic tests (or focused screening for a small set of disorders):
- Tissue biopsy or necropsy: liver, muscle, brain, bone marrow
- Skin biopsy and fibroblast cultivation for specific enzyme testing
- Specific DNA testing
A 2015 review reported that even with all these diagnostic tests, there are cases when “biochemical testing, gene sequencing, and enzymatic testing can neither confirm nor rule out an IEM, resulting in the need to rely on the patient’s clinical course.” [2]
Treatment
In the middle of the 20th century the principal treatment for some of the amino acid disorders was restriction of dietary protein and all other care was simply management of complications. In the past twenty years, enzyme replacement, gene therapy, and organ transplantation have become available and beneficial for many previously untreatable disorders. Some of the more common or promising therapies are listed:
- Dietary restriction
- E.g., reduction of dietary protein remains a mainstay of treatment for phenylketonuria and other amino acid disorders
- Dietary supplementation or replacement
- E.g., oral ingestion of cornstarch several times a day helps prevent people with glycogen storage diseases from becoming seriously hypoglycemic.
- Vitamins
- E.g., thiamine supplementation benefits several types of disorders that cause lactic acidosis.
- Intermediary metabolites, compounds, or drugs that facilitate or retard specific metabolic pathways
- Dialysis
- Enzyme replacement E.g. Acid-alpha glucosidase for Pompe disease
- Gene therapy
- Bone marrow or organ transplantation
- Treatment of symptoms and complications
- Prenatal diagnosis
Epidemiology
In a study in British Columbia, the overall incidence of the inborn errors of metabolism were estimated to be 40 per 100,000 live births or 1 in 1,400 births,[3] overall representing more than approximately 15% of single gene disorders in the population.[3]
| Type of inborn error |
Incidence |
Disease involving amino acids (e.g. PKU), organic acids,
primary lactic acidosis, galactosemia, or a urea cycle disease |
24 per 100 000 births[3] |
1 in 4,200[3] |
| Lysosomal storage disease |
8 per 100 000 births[3] |
1 in 12,500[3] |
| Peroxisomal disorder |
~3 to 4 per 100 000 of births[3] |
~1 in 30,000[3] |
| Respiratory chain-based mitochondrial disease |
~3 per 100 000 births[3] |
1 in 33,000[3] |
| Glycogen storage disease |
2.3 per 100 000 births[3] |
1 in 43,000[3] |
References
- ^ http://www.esp.org/books/garrod/inborn-errors/facsimile/
- ^ Vernon, Hilary (Jun 2015). "Inborn Errors of Metabolism: Advances in Diagnosis and Therapy". JAMA Pediatrics.
- ^ a b c d e f g h i j k l Applegarth DA, Toone JR, Lowry RB (January 2000). "Incidence of inborn errors of metabolism in British Columbia, 1969-1996". Pediatrics 105 (1): e10. doi:10.1542/peds.105.1.e10. PMID 10617747.
External links
The National Institutes of Health offers theoffice of rare diseases, home reference, medlineplus andhealth information. The National Human Genome Research Institute hosts an information center, a section forpatients and the public and additionaleducational resources. Support groups can be found atNORD, Genetic Alliance and Orphanet. The genetic education center at the KUMC has many more useful links.
|
Pathology: Medical conditions and ICD code
|
|
|
(Disease / Disorder / Syndrome / Sequence, Symptom / Sign, Injury, etc.)
|
|
| (A/B, 001–139) |
- Infectious disease/Infection: Bacterial disease
- Viral disease
- Parasitic disease
- Protozoan infection
- Helminthiasis
- Ectoparasitic infestation
- Mycosis
- Zoonosis
|
|
(C/D,
140–239 &
279–289) |
| Cancer (C00–D48, 140–239) |
|
|
| Myeloid hematologic (D50–D77, 280–289) |
|
|
| Lymphoid immune (D80–D89, 279) |
- Immunodeficiency
- Immunoproliferative disorder
- Hypersensitivity
|
|
|
| (E, 240–278) |
- Endocrine disease
- Nutrition disorder
- Inborn error of metabolism
|
|
| (F, 290–319) |
|
|
| (G, 320–359) |
- Nervous system disease
- Neuromuscular disease
|
|
| (H, 360–389) |
|
|
| (I, 390–459) |
- Cardiovascular disease
- Heart disease
- Vascular disease
|
|
| (J, 460–519) |
- Respiratory disease
- Obstructive lung disease
- Restrictive lung disease
- Pneumonia
|
|
| (K, 520–579) |
- Oral and maxillofacial pathology
- Tooth disease
- salivary gland disease
- tongue disease
- Digestive disease
- Esophageal
- Stomach
- Enteropathy
- Liver
- Pancreatic
|
|
| (L, 680–709) |
- Skin disease
- skin appendages
- Nail disease
- Hair disease
- Sweat gland disease
|
|
| (M, 710–739) |
- Musculoskeletal disorders: Myopathy
- Arthropathy
- Osteochondropathy
|
|
| (N, 580–629) |
- Urologic disease
- Nephropathy
- Urinary bladder disease
- Male genital disease
- Breast disease
- Female genital disease
|
|
| (O, 630–679) |
- Complications of pregnancy
- Obstetric labor complication
- Puerperal disorder
|
|
| (P, 760–779) |
|
|
| (Q, 740–759) |
|
|
| (R, 780–799) |
|
|
| (S/T, 800–999) |
- Bone fracture
- Joint dislocation
- Sprain
- Strain
- Subluxation
- Head injury
- Chest trauma
- Poisoning
|
|
|
Inborn error of carbohydrate metabolism: monosaccharide metabolism disorders (including glycogen storage diseases) (E73–E74, 271)
|
|
Sucrose, transport
(extracellular) |
| Disaccharide catabolism |
- Lactose intolerance
- Sucrose intolerance
|
|
| Monosaccharide transport |
- Glucose-galactose malabsorption
- Inborn errors of renal tubular transport (Renal glycosuria)
- Fructose malabsorption
|
|
|
| Hexose → glucose |
| Monosaccharide catabolism |
| fructose: |
- Essential fructosuria
- Fructose intolerance
|
|
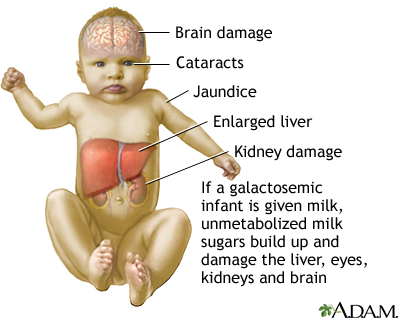
| galactose/galactosemia: |
- GALK deficiency
- GALT deficiency/GALE deficiency
|
|
|
|
| Glucose ⇄ glycogen |
| Glycogenesis |
- GSD type 0, glycogen synthase
- GSD type IV, Andersen's, branching
|
|
| Glycogenolysis |
| extralysosomal: |
- GSD type V, McArdle, muscle glycogen phosphorylase/GSD type VI, Hers', liver glycogen phosphorylase
- GSD type III, Cori's, debranching
|
|
- lysosomal/LSD: GSD type II, Pompe's, glucosidase
|
|
|
|
| Glucose ⇄ CAC |
| Glycolysis |
- MODY 2/HHF3
- GSD type VII, Tarui's, phosphofructokinase
- Triosephosphate isomerase deficiency
- Pyruvate kinase deficiency
|
|
| Gluconeogenesis |
- PCD
- Fructose bisphosphatase deficiency
- GSD type I, von Gierke, glucose 6-phosphatase
|
|
|
| Pentose phosphate pathway |
- Glucose-6-phosphate dehydrogenase deficiency
- Transaldolase deficiency
|
|
| Other |
- Hyperoxaluria
- Pentosuria
- Aldolase A deficiency
|
|
|
Index of inborn errors of metabolism
|
|
| Description |
- Metabolism
- Enzymes and pathways: citric acid cycle
- pentose phosphate
- glycoproteins
- glycosaminoglycans
- phospholipid
- cholesterol and steroid
- sphingolipids
- eicosanoids
- amino acid
- urea cycle
- nucleotide
|
|
| Disorders |
- Citric acid cycle and electron transport chain
- Glycoprotein
- Proteoglycan
- Fatty-acid
- Phospholipid
- Cholesterol and steroid
- Eicosanoid
- Amino acid
- Purine-pyrimidine
- Heme metabolism
- Symptoms and signs
|
|
| Treatment |
|
|
|
|
Inborn error of amino acid metabolism (E70–E72, 270)
|
|
| K→acetyl-CoA |
|
Lysine/straight chain
|
- Glutaric acidemia type 1
- type 2
- Hyperlysinemia
- Pipecolic acidemia
- Saccharopinuria
|
|
|
Leucine
|
- 3-hydroxy-3-methylglutaryl-CoA lyase deficiency
- 3-Methylcrotonyl-CoA carboxylase deficiency
- 3-Methylglutaconic aciduria 1
- Isovaleric acidemia
- Maple syrup urine disease
|
|
|
Tryptophan
|
|
|
|
| G |
|
G→pyruvate→citrate
|
|
Glycine
|
- D-Glyceric acidemia
- Glutathione synthetase deficiency
- Sarcosinemia
- Glycine→Creatine: GAMT deficiency
- Glycine encephalopathy
|
|
|
|
G→glutamate→
α-ketoglutarate
|
|
Histidine
|
- Carnosinemia
- Histidinemia
- Urocanic aciduria
|
|
|
Proline
|
- Hyperprolinemia
- Prolidase deficiency
|
|
|
Glutamate/glutamine
|
|
|
|
|
G→propionyl-CoA→
succinyl-CoA
|
|
Valine
|
- Hypervalinemia
- Isobutyryl-CoA dehydrogenase deficiency
- Maple syrup urine disease
|
|
|
Isoleucine
|
- 2-Methylbutyryl-CoA dehydrogenase deficiency
- Beta-ketothiolase deficiency
- Maple syrup urine disease
|
|
|
Methionine
|
- Cystathioninuria
- Homocystinuria
- Hypermethioninemia
|
|
|
General BC/OA
|
- Methylmalonic acidemia
- Methylmalonyl-CoA mutase deficiency
- Propionic acidemia
|
|
|
|
G→fumarate
|
|
Phenylalanine/tyrosine
|
|
Phenylketonuria
|
- 6-Pyruvoyltetrahydropterin synthase deficiency
- Tetrahydrobiopterin deficiency
|
|
|
Tyrosinemia
|
- Alkaptonuria/Ochronosis
- Type I tyrosinemia
- Type II tyrosinemia
- Type III tyrosinemia/Hawkinsinuria
|
|
|
Tyrosine→Melanin
|
- Albinism: Ocular albinism (1)
- Oculocutaneous albinism (Hermansky–Pudlak syndrome)
- Waardenburg syndrome
|
|
|
Tyrosine→Norepinephrine
|
- Dopamine beta hydroxylase deficiency
- reverse: Brunner syndrome
|
|
|
|
|
G→oxaloacetate
|
Urea cycle/Hyperammonemia
(arginine
|
- Argininemia
- Argininosuccinic aciduria
- Carbamoyl phosphate synthetase I deficiency
- Citrullinemia
- N-Acetylglutamate synthase deficiency
- Ornithine transcarbamylase deficiency/translocase deficiency
|
|
|
|
Transport/
IE of RTT |
- Solute carrier family: Cystinuria
- Hartnup disease
- Iminoglycinuria
- Lysinuric protein intolerance
- Fanconi syndrome: Oculocerebrorenal syndrome
- Cystinosis
|
|
| Other |
- 2-Hydroxyglutaric aciduria
- Aminoacylase 1 deficiency
- Ethylmalonic encephalopathy
- Fumarase deficiency
- Trimethylaminuria
|
|
|
Index of inborn errors of metabolism
|
|
| Description |
- Metabolism
- Enzymes and pathways: citric acid cycle
- pentose phosphate
- glycoproteins
- glycosaminoglycans
- phospholipid
- cholesterol and steroid
- sphingolipids
- eicosanoids
- amino acid
- urea cycle
- nucleotide
|
|
| Disorders |
- Citric acid cycle and electron transport chain
- Glycoprotein
- Proteoglycan
- Fatty-acid
- Phospholipid
- Cholesterol and steroid
- Eicosanoid
- Amino acid
- Purine-pyrimidine
- Heme metabolism
- Symptoms and signs
|
|
| Treatment |
|
|
|
|
Inborn error of lipid metabolism: fatty-acid metabolism disorders (E71.3, 277.81–277.85)
|
|
| Synthesis |
|
|
| Degradation |
| Acyl transport |
- Carnitine
- Primary
- I
- II
- -acylcarnitine
- Adrenoleukodystrophy
|
|
| Beta oxidation |
| General |
- Acyl CoA dehydrogenase
- Short-chain
- Medium-chain
- Long-chain 3-hydroxy
- Very long-chain
- Mitochondrial trifunctional protein deficiency: Acute fatty liver of pregnancy
|
|
| Unsaturated |
- 2,4 Dienoyl-CoA reductase deficiency
|
|
| Odd chain |
|
|
| Other |
- 3-hydroxyacyl-coenzyme A dehydrogenase deficiency
|
|
|
| To acetyl-CoA |
|
|
| Aldehyde |
|
|
|
|
Index of inborn errors of metabolism
|
|
| Description |
- Metabolism
- Enzymes and pathways: citric acid cycle
- pentose phosphate
- glycoproteins
- glycosaminoglycans
- phospholipid
- cholesterol and steroid
- sphingolipids
- eicosanoids
- amino acid
- urea cycle
- nucleotide
|
|
| Disorders |
- Citric acid cycle and electron transport chain
- Glycoprotein
- Proteoglycan
- Fatty-acid
- Phospholipid
- Cholesterol and steroid
- Eicosanoid
- Amino acid
- Purine-pyrimidine
- Heme metabolism
- Symptoms and signs
|
|
| Treatment |
|
|
|
|
Inborn error of lipid metabolism: dyslipidemia (E78, 272.0–272.6)
|
|
| Hyperlipidemia |
- Hypercholesterolemia/Hypertriglyceridemia
- Lipoprotein lipase deficiency/Type Ia
- Familial apoprotein CII deficiency/Type Ib
- Familial hypercholesterolemia/Type IIa
- Combined hyperlipidemia/Type IIb
- Familial dysbetalipoproteinemia/Type III
- Familial hypertriglyceridemia/Type IV
- Xanthoma/Xanthomatosis
|
|
| Hypolipoproteinemia |
| Hypoalphalipoproteinemia/HDL |
- Lecithin cholesterol acyltransferase deficiency
- Tangier disease
|
|
| Hypobetalipoproteinemia/LDL |
- Abetalipoproteinemia
- Apolipoprotein B deficiency
- Chylomicron retention disease
|
|
|
| Lipodystrophy |
- Barraquer–Simons syndrome
|
|
| Other |
- Lipomatosis
- Adiposis dolorosa
- Lipoid proteinosis
- APOA1 familial renal amyloidosis
|
|
|
Index of inborn errors of metabolism
|
|
| Description |
- Metabolism
- Enzymes and pathways: citric acid cycle
- pentose phosphate
- glycoproteins
- glycosaminoglycans
- phospholipid
- cholesterol and steroid
- sphingolipids
- eicosanoids
- amino acid
- urea cycle
- nucleotide
|
|
| Disorders |
- Citric acid cycle and electron transport chain
- Glycoprotein
- Proteoglycan
- Fatty-acid
- Phospholipid
- Cholesterol and steroid
- Eicosanoid
- Amino acid
- Purine-pyrimidine
- Heme metabolism
- Symptoms and signs
|
|
| Treatment |
|
|
|
|
Heme metabolism disorders (E80, 277.1, 277.4)
|
|
Porphyria,
hepatic and erythropoietic
(porphyrin) |
| early mitochondrial: |
- ALAD porphyria
- Acute intermittent porphyria
|
|
| cytoplasmic: |
- Gunther disease/congenital erythropoietic porphyria
- Porphyria cutanea tarda/Hepatoerythropoietic porphyria
|
|
| late mitochondrial: |
- Hereditary coproporphyria
- Harderoporphyria
- Variegate porphyria
- Erythropoietic protoporphyria
|
|
|
Hereditary hyperbilirubinemia
(bilirubin) |
| unconjugated: |
- Gilbert's syndrome
- Crigler–Najjar syndrome
- Lucey–Driscoll syndrome
|
|
| conjugated: |
- Dubin–Johnson syndrome
- Rotor syndrome
|
|
|
|
Index of inborn errors of metabolism
|
|
| Description |
- Metabolism
- Enzymes and pathways: citric acid cycle
- pentose phosphate
- glycoproteins
- glycosaminoglycans
- phospholipid
- cholesterol and steroid
- sphingolipids
- eicosanoids
- amino acid
- urea cycle
- nucleotide
|
|
| Disorders |
- Citric acid cycle and electron transport chain
- Glycoprotein
- Proteoglycan
- Fatty-acid
- Phospholipid
- Cholesterol and steroid
- Eicosanoid
- Amino acid
- Purine-pyrimidine
- Heme metabolism
- Symptoms and signs
|
|
| Treatment |
|
|
Index of cells from bone marrow
|
|
| Description |
- Immune system
- Cells
- Physiology
- coagulation
- proteins
- granule contents
- colony-stimulating
- heme and porphyrin
|
|
| Disease |
- Red blood cell
- Monocyte and granulocyte
- Neoplasms and cancer
- Histiocytosis
- Symptoms and signs
- Blood tests
|
|
| Treatment |
- Transfusion
- Drugs
- thrombosis
- bleeding
- other
|
|
|
|
Inborn error of purine-pyrimidine metabolism (E79, 277.2)
|
|
| Purine metabolism |
|
Anabolism
|
- Adenylosuccinate lyase deficiency
- Adenosine Monophosphate Deaminase Deficiency type 1
|
|
|
Nucleotide salvage
|
- Lesch-Nyhan syndrome/Hyperuricemia
- Adenine phosphoribosyltransferase deficiency
|
|
|
Catabolism
|
- Adenosine deaminase deficiency
- Purine nucleoside phosphorylase deficiency
- Xanthinuria
- Gout
- Mitochondrial neurogastrointestinal encephalopathy syndrome
|
|
|
| Pyrimidine metabolism |
|
Anabolism
|
- Orotic aciduria
- Miller syndrome
|
|
|
Catabolism
|
- Dihydropyrimidine dehydrogenase deficiency
|
|
|
|
Index of inborn errors of metabolism
|
|
| Description |
- Metabolism
- Enzymes and pathways: citric acid cycle
- pentose phosphate
- glycoproteins
- glycosaminoglycans
- phospholipid
- cholesterol and steroid
- sphingolipids
- eicosanoids
- amino acid
- urea cycle
- nucleotide
|
|
| Disorders |
- Citric acid cycle and electron transport chain
- Glycoprotein
- Proteoglycan
- Fatty-acid
- Phospholipid
- Cholesterol and steroid
- Eicosanoid
- Amino acid
- Purine-pyrimidine
- Heme metabolism
- Symptoms and signs
|
|
| Treatment |
|
|
|
|
Inborn error of steroid metabolism
|
|
| Mevalonate pathway |
- Hyper-IgD syndrome
- Mevalonate kinase deficiency
|
|
| To cholesterol |
- 7-Dehydrocholesterol path: Hydrops-ectopic calcification-moth-eaten skeletal dysplasia
- CHILD syndrome
- Conradi-Hünermann syndrome
- Lathosterolosis
- Smith-Lemli-Opitz syndrome
- desmosterol path: Desmosterolosis
|
|
| Steroids |
Corticosteroid
(including CAH) |
- aldosterone: Glucocorticoid remediable aldosteronism
- cortisol/cortisone: CAH 17α hydroxylase
- CAH 11β hydroxylase
- both: CAH 3β dehydrogenase
- CAH 21α hydroxylase
- Apparent mineralocorticoid excess syndrome/11β dehydrogenase
|
|
| Sex steroid |
| To androgens |
- 17-beta-hydroxysteroid dehydrogenase deficiency
- 5-alpha-reductase deficiency
- Pseudovaginal perineoscrotal hypospadias
|
|
| To estrogens |
- Aromatase deficiency
- Aromatase excess syndrome
|
|
|
| Other |
- X-linked ichthyosis
- Antley-Bixler syndrome
|
|
|
|
Index of inborn errors of metabolism
|
|
| Description |
- Metabolism
- Enzymes and pathways: citric acid cycle
- pentose phosphate
- glycoproteins
- glycosaminoglycans
- phospholipid
- cholesterol and steroid
- sphingolipids
- eicosanoids
- amino acid
- urea cycle
- nucleotide
|
|
| Disorders |
- Citric acid cycle and electron transport chain
- Glycoprotein
- Proteoglycan
- Fatty-acid
- Phospholipid
- Cholesterol and steroid
- Eicosanoid
- Amino acid
- Purine-pyrimidine
- Heme metabolism
- Symptoms and signs
|
|
| Treatment |
|
|
Index of hormones
|
|
| Description |
- Glands
- Hormones
- thyroid
- mineralocorticoids
- Physiology
- Development
|
|
| Disease |
- Diabetes
- Congenital
- Neoplasms and cancer
- Other
- Symptoms and signs
|
|
| Treatment |
- Procedures
- Drugs
- calcium balance
- corticosteroids
- oral hypoglycemics
- pituitary and hypothalamic
- thyroid
|
|
|
|
Metabolic disorders of vitamins, coenzymes, and cofactors
|
|
| B7 Biotin/MCD |
- Biotinidase deficiency
- Holocarboxylase synthetase deficiency
|
|
| Other B |
- B5 (Pantothenate kinase-associated neurodegeneration)
- B12 (Methylmalonic acidemia)
|
|
| Other vitamin |
- Familial isolated vitamin E deficiency
|
|
| Nonvitamin cofactor |
- Tetrahydrobiopterin deficiency
- Molybdenum cofactor deficiency
|
|
|
Index of nutrition
|
|
| Description |
- Vitamins
- Cofactors
- Metal metabolism
- Fats
- metabolism
- intermediates
- lipoproteins
- Sugars
- Glycolysis
- Glycogenesis and glycogenolysis
- Fructose and galactose
|
|
| Disease |
- Vitamins
- Carbohydrate
- Lipid
- Metals
- Other
- Symptoms and signs
- Tests
|
|
| Treatment |
- Drugs
- Vitamins
- Mineral supplements
|
|
|