WordNet
- an impairment of health or a condition of abnormal functioning
- caused by or altered by or manifesting disease or pathology; "diseased tonsils"; "a morbid growth"; "pathologic tissue"; "pathological bodily processes" (同)morbid, pathologic, pathological
PrepTutorEJDIC
- (体の)『病気』,疾患 / (精神・道徳などの)病気,病弊
- 女性の話術芸人 =diseur
- 病気にかかった / 病的な,不健全な(morbid)
Wikipedia preview
出典(authority):フリー百科事典『ウィキペディア(Wikipedia)』「2013/12/22 19:54:04」(JST)
wiki en
This article is about the specific disease caused by a defect in the SMN1 gene. For a list of similarly-named conditions caused by defects in other genes, see Spinal muscular atrophies.
| Spinal muscular atrophy | |
|---|---|
| Classification and external resources | |
Location of neurons affected by spinal muscular atrophy in the spinal cord
|
|
| ICD-10 | G12.0-G12.1 |
| ICD-9 | 335.0-335.1 |
| OMIM | 253300 253550 253400 271150 |
| DiseasesDB | 14093 32911 12315 34537 |
| MedlinePlus | 000996 |
| eMedicine | Spinal Muscular Atrophy Spinal Muscle Atrophy |
| MeSH | D014897 |
| GeneReviews |
|
Spinal muscular atrophy (SMA) is an autosomal recessive disease caused by a genetic defect in the SMN1 gene, which encodes SMN, a protein widely expressed in all eukaryotic cells. SMN1 is apparently selectively necessary for survival of motor neurons, as diminished abundance of the protein results in death of neuronal cells in the anterior horn of the spinal cord and subsequent system-wide muscle wasting (atrophy).
Spinal muscular atrophy manifests in various degrees of severity which all have in common general muscle wasting and mobility impairment. Other body systems may be affected as well, particularly in early-onset forms. SMA is the most common genetic cause of infant death.
The term spinal muscular atrophy is used as both a specific term for the genetic disorder caused by deficient SMN, and a general label for a larger number of rare disorders having in common a genetic cause and slow progression of weakness without sensory impairment caused by disease of motor neurons in the spinal cord and brainstem – see spinal muscular atrophies for a comparison chart.
Contents
- 1 Signs and symptoms
- 2 Causes
- 3 Diagnosis
- 3.1 Types
- 4 Treatment
- 4.1 Palliative care
- 5 Prognosis
- 6 Research directions
- 7 See also
- 8 References
- 9 External links
Signs and symptoms[edit]
The symptoms vary greatly depending on the SMA type involved, the stage of the disease and individual factors and commonly include:
- Areflexia, particularly in extremities
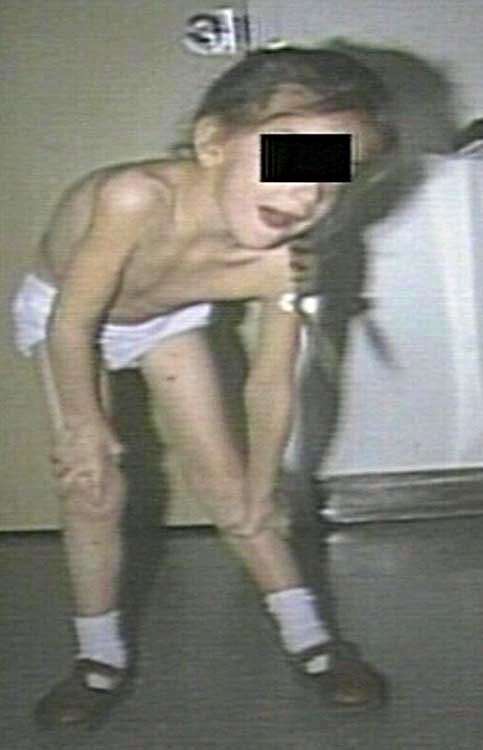
- Overall muscle weakness, poor muscle tone, limpness or a tendency to flop (the "floppy baby" syndrome)
- Difficulty achieving developmental milestones, difficulty sitting/standing/walking
- In infants: adopting of a frog-leg position when sitting (hips abducted and knees flexed)
- Loss of strength of the respiratory muscles: weak cough, weak cry (infants), accumulation of secretions in the lungs or throat, respiratory distress
- Bell-shaped torso (caused by using only abdominal muscles for respiration)
- Clenched fists with sweaty hands
- Head often tilted to one side, even when lying down
- Fasciculations (twitching) of the tongue
- Difficulty sucking or swallowing, poor feeding
- Arthrogryposis (multiple congenital contractures)
- Weight lower than normal
Causes[edit]
Spinal muscular atrophy has an autosomal recessive pattern of inheritance.
Spinal muscular atrophy is linked to a genetic mutation in the SMN1 gene.[1]
Human chromosome 5 contains two nearly identical genes at location 5q13: a telomeric copy SMN1 and a centromeric copy SMN2. In healthy individuals, the SMN1 gene codes the survival of motor neuron protein (SMN) which, as its name says, plays a crucial role in survival of motor neurons. The SMN2 gene, on the other hand - due to a variation in a single nucleotide (840.C→T) - undergoes alternative splicing at the junction of intron 6 to exon 8, with only 10-20% of SMN2 transcripts coding a fully functional survival of motor neuron protein (SMN-fl) and 80-90% of transcripts resulting in a truncated protein compound (SMNΔ7) which is rapidly degraded in the cell.
In SMA-affected individuals, the SMN1 gene is mutated in such a way that it is unable to correctly code the SMN protein - due to either a deletion occurring at exon 7 or to other point mutations (frequently resulting in the functional conversion of the SMN1 sequence into SMN2). All patients, however, retain at least one copy of the SMN2 gene (with most having 2-4 of them) which still code small amounts of SMN protein - around 10-20% of the normal level - allowing neurons to survive. In the long run, however, reduced availability of the SMN protein results in gradual death of motor neuron cells in the anterior horn of spinal cord and the brain. Consequently, motor muscles undergo progressive atrophy.
Muscles of lower extremities are usually affected first, followed by muscles of upper extremities, spine and neck and, in more severe cases, pulmonary and mastication muscles. Proximal muscles are always affected earlier and in a greater degree than distal.
The severity of SMA symptoms is broadly related to how well the remaining SMN2 genes can make up for the loss of SMN1. This is partly related to the number of SMN2 gene copies present on the chromosome. Whilst healthy individuals carry two SMN2 gene copies, SMA patients can have anything between 1 and 4 (or more) of them, with the greater the number of SMN2 copies the milder the disease severity. Thus, most SMA type I babies have one or two SMN2 copies; SMA II and III patients usually have at least three SMN2 copies; and SMA IV patients normally have at least four of them. However, the correlation between symptom severity and SMN2 copy number is not absolute and there seem to exist other factors impacting on the disease phenotype.[2]
Spinal muscular atrophy is inherited in an autosomal recessive pattern, which means that the defective gene is located on an autosome, and two copies of the defective gene - one from each parent - are required to inherit the disorder: the parents do not need to be themselves affected. SMA seems to appear de novo (i.e., without any hereditary causes) in around 2-4% of cases.
Spinal muscular atrophy affects individuals of all races, unlike other well known autosomal recessive disorders like sickle cell disease and cystic fibrosis which have significant differences in occurrence rate between races. The overall incidence of SMA, of all types and across all ethnic groups, is in the range of 1 per 10,000 individuals; the gene frequency is thus around 1:100, therefore, approximately one in 50 persons are carriers.[3][4] There are no known health consequences of being a carrier, and presently the only way one may know to consider the possibility is if a relative is affected.
Finally, there are also reports of occurrence of SMA type I and SMA type II in the same sibship together; scientific explanation of this phenomena (intrafamilial variability) have been advanced by the italian researcher of the CNR (The National Rersearch Council of Italy) Enrico Parano, who has suggested that these cases might be due to additional de novo deletion of the SMN gene, not involving the NAIP gene (94).
Diagnosis[edit]
Prenatal screening is controversial, because of its cost on the one hand, and the severity of the disease on the other hand. Some researchers have concluded that population screening for SMA is not cost-effective, at a cost of $5 million per case averted in USA.[5] Others conclude that SMA meets the criteria for screening programs and relevant testing should be offered to all couples.[6]
Very severe SMA (type 0/I) can be sometimes evident before birth - reduction in fetal movement in the final months of pregnancy; else, it manifests within the first few weeks or months of life when abnormally low muscle tone is observed (the "floppy baby syndrome").
Further, for all SMA types,
- Patient will present hypotonia associated with absent reflexes
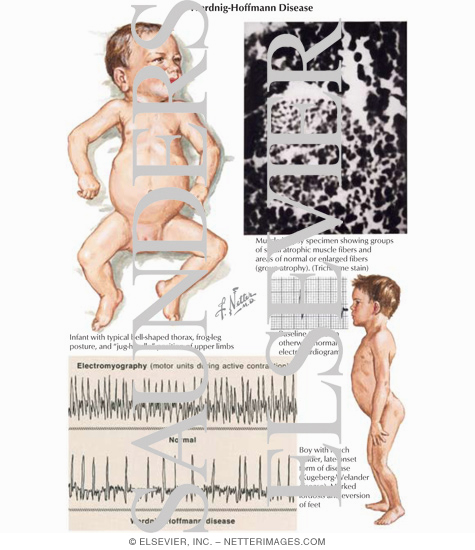
- Electromyogram will show fibrillation and muscle denervation[7]
- Serum creatine kinase may be normal or increased[citation needed]
- Genetic testing will show bi-allelic deletion of exon 7 of the SMN1 gene – this is conclusive of the disease.
Types[edit]
SMA manifests over a wide range of severity affecting infants through adults. The disease spectrum is variously divided into 3–5 types, in accordance either with the age of onset of symptoms or with the highest attained milestone of motor development.
The most commonly used classification is as follows:
| Type | Eponym | Usual age of onset | Characteristics | OMIM |
|---|---|---|---|---|
| I: Infantile | Werdnig–Hoffmann disease | 0–6 months | The severe form manifests in the first months of life, usually with a quick and unexpected onset ("floppy baby syndrome"). Rapid motor neuron death causes inefficiency of the major bodily organs - especially of the respiratory system - and pneumonia-induced respiratory failure is the most frequent cause of death. Babies diagnosed with SMA type I do not generally live past two years of age, with death occurring as early as within weeks in the most severe cases (sometimes termed SMA type 0). With proper respiratory support, those with milder SMA type I phenotypes, which account for around 10% of cases, are known to live into adolescence and adulthood. | 253300 |
| II: Intermediate | Dubowitz disease | 6–18 months | The intermediate form affects children who are never able to stand and walk but who are able to maintain a sitting position at least some time in their life. The onset of weakness is usually noticed some time between 6 and 18 months. The progress is known to vary greatly, some patients gradually grow weaker over time while others through careful maintenance avoid any progression. Body muscles are weakened, and the respiratory system is a major concern. Life expectancy is somewhat reduced but most SMA II patients live well into adulthood. | 253550 |
| III: Juvenile | Kugelberg–Welander disease | >18 months | The juvenile form usually manifests after 18 months of age and describes patients who are able to walk without support at some time, although many later lose this ability. Respiratory involvement is less noticeable, and life expectancy is normal or near normal. | 253400 |
| IV: Adult-onset | Adulthood | The adult-onset form (sometimes classified as a late-onset SMA type III) usually manifests after the third decade of life with gradual weakening of muscles – mainly affects proximal muscles of the extremities – frequently rendering the patient wheelchair-bound. Other complications are rare, and life expectancy is unaffected. | 271150 |
The most severe form of SMA type I is sometimes termed SMA type 0 (or severe infantile SMA) and is diagnosed in babies that are born so weak that they can survive only a few weeks even with intensive respiratory support. SMA type 0 should not be confused with SMARD1 which may have very similar symptoms and course but has a different genetic cause than SMA.
Development milestone attainment is commonly measured using a specially modified Hammersmith Functional Motor Scale.[8][9][10][11]
The eponymous label Werdnig-Hoffmann disease (often misspelled with a single "n") refers to the earliest clinical descriptions of childhood SMA by Johann Hoffmann and Guido Werdnig. The eponymous term Kugelberg-Welander disease after Erik Klas Hendrik Kugelberg (1913-1983) and Lisa Welander (1909-2001), who distinguished SMA from muscular dystrophy.[12] Rarely used Dubowitz disease (not to be confused with Dubowitz syndrome) is named after Victor Dubowitz, an English neurologist who authored several studies on the intermediate SMA phenotype.
Treatment[edit]
There is no known cure for spinal muscular atrophy.
Palliative care[edit]
Care is symptomatic. Main areas of concern are as follows:
- Orthopaedics — Weak spine muscles may lead to development of kyphosis, scoliosis and other orthopaedic problems. Spine fusion is sometimes performed in SMA I/II patients once they reach the age of 8-10 to relieve the pressure of a deformed spine on the lungs. SMA patients might also benefit greatly from various forms of physiotherapy and occupational therapy.
- Respiratory care — Respiratory system requires utmost attention in SMA as once weakened it never fully recovers. Weakened pulmonary muscles in SMA type I/II patients can make breathing more difficult and pose a risk of hypoxiation, especially in sleep when muscles are more relaxed. Impaired cough reflex poses a constant risk of respiratory infection and pneumonia. Non-invasive ventilation (BiPAP) is frequently used and tracheostomy may be sometimes performed in more severe cases;[13] both methods of ventilation prolong survival in a comparable degree, although tracheostomy prevents speech development.[14]
- Nutritional care — Difficulties in jaw opening, chewing and swallowing food might pose SMA patients at risk of malnutrition. A feeding tube can be necessary in SMA type I and more severe type II patients.[15][16][17] Additionally, metabolic abnormalities resulting from SMA impair β-oxidation of fatty acids in muscles and can lead to organic acidemia and consequent muscle damage, especially when fasting.[18][19] It is suggested that SMA patients, especially those with more severe forms of disease, reduce intake of fat and avoid prolonged fasting (i.e., eat more frequently than healthy people).[20]
- Mobility — Assistive technologies may help in managing movement and daily activity and greatly increase the quality of life.
- Cardiology — Although heart is not a matter of routine concern, a link between SMA and certain heart conditions has been suggested.[21][22][23][24]
- Mental health — SMA children do not differ from the general population in their behaviour; their cognitive development can be slightly faster, and certain aspects of their intelligence are above the average.[25][26][27] Despite their disability, SMA-affected people report high degree of satisfaction from life.[28]
Palliative care in SMA has been standardised in the Consensus Statement for Standard of Care in Spinal Muscular Atrophy which has been recommended for standard adoption worldwide.
Prognosis[edit]
Generally, patients tend to deteriorate over time, but prognosis varies with the SMA type and disease progress which shows a great degree of individual variability.
The majority of children diagnosed with SMA type 0/I do not reach the age of 10, recurrent respiratory problems being the primary cause of morbidity.[29] With proper care, milder SMA type I cases have lived into adulthood.[30]
In SMA type II, the course of the disease is stable or slowly progressing and life expectancy is somewhat reduced compared to the healthy population, although patients usually live to become parents and grandparents.
SMA type III has normal or nearly normal life expectancy if standards of care are followed. Adult-onset SMA usually means only mobility impairment and does not affect life expectancy.
Research directions[edit]
Since the underlying genetic mechanism of SMA was described in 1990, several therapeutic approaches have been proposed and investigated. Since a vast number of in vitro and animal modelling studies suggest that restoration of SMN levels reverts SMA symptoms, the majority of emerging therapies focus on increasing the availability of SMN protein to motor neurons.
The main therapeutic pathways under research as of December 2011 include:[31][32][33][34][35][36][37][38][39]
- Gene therapy — aims at correcting the SMN1 gene function through inserting specially crafted nucleotide sequences with the help of a viral vector.[40] In the context of SMA, it is currently being researched using the scAAV9 viral vector at the Ohio State University and Nationwide Children's Hospital, USA, and the University of Sheffield, United Kingdom, as well as by Genzyme Corporation, USA, and Généthon, France. In one study this method has resulted in the greatest survival increase achieved to-date in a SMNΔ7 mouse model (median survival of 400 days in treated mice as opposed to 15 days in untreated mice).[citation needed] Safety and pharmacokinetics of scAAV9 viral vector has been tested in non-human primates.[41]
- Stem cell therapy — aims at offering protection to affected neurons through injection of specially prepared human stem cells in the spinal cord which subsequently develop into neuronal cells able to code full-length SMN protein, and is developed commercially in the context of SMA by California Stem Cell, USA. Experimental stem cell therapy is also offered to SMA patients - based on limited research and with unclear outcome - in private clinics in Brazil, China, Russia, Ukraine and Lebanon.
- SMN2 activation — aims at increasing expression of the SMN2 gene and thus increasing the amount of full-length SMN available; compounds under investigation include:
-
- Growth hormone
- Histone deacetylase inhibitors:[42]
-
- Aliphatic compounds:
-
- Butyrates: sodium butyrate and sodium phenylbutyrate — promising in vitro and demonstrated effective in mouse models,[43][44][45] proved ineffective in symptomatic SMA patients (probably due to extremely short half-life),[46] still being trialled in pre-symptomatic type I/II infants[47]
- Valproic acid — formerly used widely on experimental basis due to earlier research showing its effectiveness in vitro[48] and in mouse models,[49] in achievable concentrations demonstrated ineffective in SMA patients[50][51][52] and even shown to aggravate SMA symptoms[53]
- Benzamides:
-
- M344 — shown very effective in mouse models,[54] so far not trialled in SMA patients
- Hydroxamic acids:
-
- CBHA, SBHA — shown very promising in vitro
- Entinostat (MS-275) — shown very promising in vitro
- Panobinostat (LBH-589) — shown very effective in mouse models,[55] not trialled in SMA patients due to toxicity at required dosage
- Trichostatin A — shown effective in mouse models,[56][57] so far not trialled in SMA patients
- Vorinostat (SAHA) — shown effective in mouse models,[58] so far not trialled in SMA patients
- Hydroxycarbamide (hydroxyurea) — shown effective in mouse models[59] and subsequently commercially researched by Novo Nordisk, Denmark, but demonstrated no effect on SMA patients in subsequent clinical trials[60]
- Natural polyphenol compounds: resveratrol, curcumin — moderate effectiveness on muscle strength supported by anecdotal evidence from patients and limited research in vitro[61][62]
- Prolactin — recently shown effective in mouse models,[63] so far not trialled in SMA patients
- Salbutamol (albuterol) — demonstrated moderately effective in vitro[64] and in two clinical trials involving SMA II/III patients[65][66]
- SMN2 alternative splicing modulation — targets the alternative splicing of the SMN2 gene so as to achieve a higher proportion of full-length SMN transcripts (sometimes called "gene conversion SMN2→SMN1"); compounds under investigation include:
-
- Aclarubicin — shown effective in human cells from type I SMA patients,[67] not trialled any further due to toxicity at required dosage
- Antisense oligonucleotides:[68][69][70]
-
- ISIS-SMNx — a proprietary molecule under development by Isis Pharmaceuticals, USA, and as of July 2012, posed for a phase II clinical trial; has Fast Track Designation in the USA and Orphan Medicinal Product Recommendation in the European Union
- PTK-SMA1 — a proprietary small molecule splicing modulator of the tetracyclines group under development by Paratek Pharmaceutical, USA
- Quinazolines:[71]
-
- RG3039 (formerly, Quinazoline495) — a proprietary quinazoline derivative under development by Repligen Corporation, USA, and as of July 2012, scheduled for phase II clinical trial; has an Orphan Drug Designation and Fast Track Designation in the USA and Orphan Medicinal Product Recommendation in the European Union
- Sodium orthovanadate — shown to modulate alternative splicing in one study in vitro[72]
- SMN stabilisation — aims at stabilising the SMNΔ7 protein (the short-lived defective protein coded by the SMN2 gene) so that it is able to sustain neuronal cells;[73] investigated compounds include:
-
- Aminoglycosides — shown to increase SMN protein availability[74][75]
-
- TC-007 — a proprietary aminoglycoside antibiotic under commercial development by Tikvah Therapeutics, USA
- Indoprofen[76]
- Neuroprotection — aims at prolonging survival of motor neurons even with low levels of SMN; investigated compounds include:
-
- β-lactam antibiotics (e.g., ceftriaxone) — shown promising in vitro[77][78]
- Follistatin — shown promising in vitro[79]
- Olesoxime — a proprietary compound developed by a French company Trophos, currently (2011-2013) under a phase II clinical trial in USA and Europe
- Riluzole — a compound approved for treatment of ALS, currently being investigated for SMA at University of Angers, France
- Thyrotropin-releasing hormone (TRH) — shown promising in vitro and in open-label uncontrolled clinical trials[80][81][82] yet did not prove effective in double-blind placebo-controlled trials[35]
- An unclear mechanism of action is found in the following compounds currently under research:
-
- PTC-X — three proprietary compounds under joint development by PTC Therapeutics, USA, and Hoffmann-La Roche, Switzerland[83]
- RE-003 — a compound being developed by Retrophin.[84]
In vivo research is usually conducted using genetically engineered Caenorhabditis elegans,[85][86] Drosophila,[85][87][88] zebrafish[89] and mouse[90] models; larger animal models are under development.[91] SMA patients can have a chance of participating in research by entering their details into international SMA patient registries. A list of clinical trials targeting SMA can be found here [1].
It has to be noted, though, that SMA therapeutics seem to be most effective when given immediately after birth, then losing their efficacy with the patient's age. This might be related to the variation in time of the needs for SMN protein by neuronal cells. However, this also poses a major therapeutic problem as hardly ever is SMA diagnosed at birth.[92][93]
See also[edit]
- SMA Treatment Acceleration Act
- Spinal muscular atrophies
- Motor neuron disease
- Survival of motor neuron
- Spinal muscular atrophy with respiratory distress type 1
- Spinal and bulbar muscular atrophy
References[edit]
- ^ Brzustowicz, L. M.; Lehner, T.; Castilla, L. H.; Penchaszadeh, G. K.; Wilhelmsen, K. C.; Daniels, R.; Davies, K. E.; Leppert, M.; Ziter, F.; Wood, D.; Dubowitz, V.; Zerres, K.; Hausmanowa-Petrusewicz, I.; Ott, J.; Munsat, T. L.; Gilliam, T. C. (1990). "Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2–13.3". Nature 344 (6266): 540–541. doi:10.1038/344540a0. PMID 2320125. edit
- ^ Jędrzejowska, M.; Milewski, M.; Zimowski, J.; Borkowska, J.; Kostera-Pruszczyk, A.; Sielska, D.; Jurek, M.; Hausmanowa-Petrusewicz, I. (2009). "Phenotype modifiers of spinal muscular atrophy: The number of SMN2 gene copies, deletion in the NAIP gene and probably gender influence the course of the disease". Acta Biochimica Polonica 56 (1): 103–108. PMID 19287802. edit
- ^ Su, Y. N.; Hung, C. C.; Lin, S. Y.; Chen, F. Y.; Chern, J. P. S.; Tsai, C.; Chang, T. S.; Yang, C. C.; Li, H.; Ho, H. N.; Lee, C. N. (2011). "Carrier Screening for Spinal Muscular Atrophy (SMA) in 107,611 Pregnant Women during the Period 2005–2009: A Prospective Population-Based Cohort Study". In Schrijver, Iris. PLoS ONE 6 (2): e17067. doi:10.1371/journal.pone.0017067. PMC 3045421. PMID 21364876. edit
- ^ Sugarman, E. A.; Nagan, N.; Zhu, H.; Akmaev, V. R.; Zhou, Z.; Rohlfs, E. M.; Flynn, K.; Hendrickson, B. C.; Scholl, T.; Sirko-Osadsa, D. A.; Allitto, B. A. (2011). "Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of >72 400 specimens". European Journal of Human Genetics 20 (1): 27–32. doi:10.1038/ejhg.2011.134. PMC 3234503. PMID 21811307. edit
- ^ Little, S. E.; Janakiraman, V.; Kaimal, A.; Musci, T.; Ecker, J.; Caughey, A. B. (2010). "The cost-effectiveness of prenatal screening for spinal muscular atrophy". American Journal of Obstetrics and Gynecology 202 (3): 253.2e1. doi:10.1016/j.ajog.2010.01.032. PMID 20207244. edit
- ^ Prior, T. W.; Professional Practice Guidelines Committee (2008). "Carrier screening for spinal muscular atrophy". Genetics in Medicine 10 (11): 840–842. doi:10.1097/GIM.0b013e318188d069. PMC 3110347. PMID 18941424. edit
- ^ Rutkove, S. B.; Shefner, J. M.; Gregas, M.; Butler, H.; Caracciolo, J.; Lin, C.; Fogerson, P. M.; Mongiovi, P.; Darras, B. T. (2010). "Characterizing spinal muscular atrophy with electrical impedance myography". Muscle & Nerve 42 (6): 915. doi:10.1002/mus.21784. edit
- ^ Main, M.; Kairon, H.; Mercuri, E.; Muntoni, F. (2003). "The Hammersmith Functional Motor Scale for Children with Spinal Muscular Atrophy: A Scale to Test Ability and Monitor Progress in Children with Limited Ambulation". European Journal of Paediatric Neurology 7 (4): 155–159. doi:10.1016/S1090-3798(03)00060-6. PMID 12865054. edit
- ^ Krosschell, K. J.; Maczulski, J. A.; Crawford, T. O.; Scott, C.; Swoboda, K. J. (2006). "A modified Hammersmith functional motor scale for use in multi-center research on spinal muscular atrophy". Neuromuscular Disorders 16 (7): 417–426. doi:10.1016/j.nmd.2006.03.015. PMID 16750368. edit
- ^ O'Hagen, J. M.; Glanzman, A. M.; McDermott, M. P.; Ryan, P. A.; Flickinger, J.; Quigley, J.; Riley, S.; Sanborn, E.; Irvine, C.; Martens, W. B.; Annis, C.; Tawil, R.; Oskoui, M.; Darras, B. T.; Finkel, R. S.; De Vivo, D. C. (2007). "An expanded version of the Hammersmith Functional Motor Scale for SMA II and III patients". Neuromuscular Disorders 17 (9–10): 693–697. doi:10.1016/j.nmd.2007.05.009. PMID 17658255. edit
- ^ Glanzman, A. M.; O'Hagen, J. M.; McDermott, M. P.; Martens, W. B.; Flickinger, J.; Riley, S.; Quigley, J.; Montes, J.; Dunaway, S.; Deng, L.; Chung, W. K.; Tawil, R.; Darras, B. T.; De Vivo, D. C.; Kaufmann, P.; Finkel, R. S.; Pediatric Neuromuscular Clinical Research Network for Spinal Muscular Atrophy (PNCR) (2011). "Validation of the Expanded Hammersmith Functional Motor Scale in Spinal Muscular Atrophy Type II and III". Journal of Child Neurology 26 (12): 1499–1507. doi:10.1177/0883073811420294. PMID 21940700. edit
- ^ Dubowitz, V. (2009). "Ramblings in the history of spinal muscular atrophy". Neuromuscular Disorders 19 (1): 69–73. doi:10.1016/j.nmd.2008.10.004. PMID 18951794. edit
- ^ Bach, J. R.; Niranjan, V.; Weaver, B. (2000). "Spinal Muscular Atrophy Type 1: A Noninvasive Respiratory Management Approach". Chest 117 (4): 1100–1105. doi:10.1378/chest.117.4.1100. PMID 10767247. edit
- ^ Bach, J. R.; Saltstein, K.; Sinquee, D.; Weaver, B.; Komaroff, E. (2007). "Long-Term Survival in Werdnig–Hoffmann Disease". American Journal of Physical Medicine & Rehabilitation 86 (5): 339. doi:10.1097/PHM.0b013e31804a8505. PMID 17449977. edit
- ^ Messina, S.; Pane, M.; De Rose, P.; Vasta, I.; Sorleti, D.; Aloysius, A.; Sciarra, F.; Mangiola, F.; Kinali, M.; Bertini, E.; Mercuri, E. (2008). "Feeding problems and malnutrition in spinal muscular atrophy type II". Neuromuscular Disorders 18 (5): 389–393. doi:10.1016/j.nmd.2008.02.008. PMID 18420410. edit
- ^ Chen, Y. S.; Shih, H. H.; Chen, T. H.; Kuo, C. H.; Jong, Y. J. (2011). "Prevalence and Risk Factors for Feeding and Swallowing Difficulties in Spinal Muscular Atrophy Types II and III". The Journal of Pediatrics. doi:10.1016/j.jpeds.2011.08.016. edit
- ^ Tilton, A.; Miller, M.; Khoshoo, V. (1998). "Nutrition and swallowing in pediatric neuromuscular patients". Seminars in Pediatric Neurology 5 (2): 106–115. doi:10.1016/S1071-9091(98)80026-0. PMID 9661244. edit
- ^ Tein, I.; Sloane, A. E.; Donner, E. J.; Lehotay, D. C.; Millington, D. S.; Kelley, R. I. (1995). "Fatty acid oxidation abnormalities in childhood-onset spinal muscular atrophy: Primary or secondary defect(s)?". Pediatric neurology 12 (1): 21–30. doi:10.1016/0887-8994(94)00100-G. PMID 7748356. edit
- ^ Crawford, T. O.; Sladky, J. T.; Hurko, O.; Besner-Johnston, A.; Kelley, R. I. (1999). "Abnormal fatty acid metabolism in childhood spinal muscular atrophy". Annals of Neurology 45 (3): 337–343. doi:10.1002/1531-8249(199903)45:3<337::AID-ANA9>3.0.CO;2-U. PMID 10072048. edit
- ^ Leighton, S. (2003). "Nutrition issues associated with spinal muscular atrophy". Nutrition & Dietetics 60 (2): 92–96.
- ^ Rudnik-Schoneborn, S.; Heller, R.; Berg, C.; Betzler, C.; Grimm, T.; Eggermann, T.; Eggermann, K.; Wirth, R.; Wirth, B.; Zerres, K. (2008). "Congenital heart disease is a feature of severe infantile spinal muscular atrophy". Journal of Medical Genetics 45 (10): 635–638. doi:10.1136/jmg.2008.057950. PMID 18662980. edit
- ^ Heier, C. R.; Satta, R.; Lutz, C.; Didonato, C. J. (2010). "Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice". Human Molecular Genetics 19 (20): 3906–3918. doi:10.1093/hmg/ddq330. PMC 2947406. PMID 20693262. edit
- ^ Shababi, M.; Habibi, J.; Yang, H. T.; Vale, S. M.; Sewell, W. A.; Lorson, C. L. (2010). "Cardiac defects contribute to the pathology of spinal muscular atrophy models". Human Molecular Genetics 19 (20): 4059–4071. doi:10.1093/hmg/ddq329. PMID 20696672. edit
- ^ Bevan, A. K.; Hutchinson, K. R.; Foust, K. D.; Braun, L.; McGovern, V. L.; Schmelzer, L.; Ward, J. G.; Petruska, J. C.; Lucchesi, P. A.; Burghes, A. H. M.; Kaspar, B. K. (2010). "Early heart failure in the SMNΔ7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery". Human Molecular Genetics 19 (20): 3895–3905. doi:10.1093/hmg/ddq300. PMC 2947399. PMID 20639395. edit
- ^ Von Gontard, A.; Zerres, K.; Backes, M.; Laufersweiler-Plass, C.; Wendland, C.; Melchers, P.; Lehmkuhl, G.; Rudnik-Schöneborn, S. (2002). "Intelligence and cognitive function in children and adolescents with spinal muscular atrophy". Neuromuscular Disorders 12 (2): 130–136. doi:10.1016/S0960-8966(01)00274-7. PMID 11738354. edit
- ^ Billard, C.; Gillet, P.; Signoret, J. L.; Uicaut, E.; Bertrand, P.; Fardeau, M.; Barthez-Carpentier, M. A.; Santini, J. J. (1992). "Cognitive functions in duchenne muscular dystrophy: A reappraisal and comparison with spinal muscular atrophy". Neuromuscular Disorders 2 (5–6): 371–378. doi:10.1016/S0960-8966(06)80008-8. PMID 1300185. edit
- ^ Laufersweiler-Plass, C.; Rudnik-Schöneborn, S.; Zerres, K.; Backes, M.; Lehmkuhl, G.; Von Gontard, A. (2002). "Behavioural problems in children and adolescents with spinal muscular atrophy and their siblings". Developmental Medicine & Child Neurology 45. doi:10.1017/S0012162203000082. edit
- ^ De Oliveira, C. M.; Araújo, A. P. D. Q. C. (2011). "Self-reported quality of life has no correlation with functional status in children and adolescents with spinal muscular atrophy". European Journal of Paediatric Neurology 15 (1): 36–39. doi:10.1016/j.ejpn.2010.07.003. PMID 20800519. edit
- ^ Yuan, N.; Wang, C. H.; Trela, A.; Albanese, C. T. (2007). "Laparoscopic Nissen Fundoplication During Gastrostomy Tube Placement and Noninvasive Ventilation May Improve Survival in Type I and Severe Type II Spinal Muscular Atrophy". Journal of Child Neurology 22 (6): 727–731. doi:10.1177/0883073807304009. PMID 17641258. edit
- ^ Bach, J. R. (2007). "Medical Considerations of Long-Term Survival of Werdnig–Hoffmann Disease". American Journal of Physical Medicine & Rehabilitation 86 (5): 349. doi:10.1097/PHM.0b013e31804b1d66. PMID 17449979. edit
- ^ Pruss, R. M.; Giraudon-Paoli, M.; Morozova, S.; Berna, P.; Abitbol, J. L.; Bordet, T. (2010). "Drug discovery and development for spinal muscular atrophy: Lessons from screening approaches and future challenges for clinical development". Future Medicinal Chemistry 2 (9): 1429–1440. doi:10.4155/FMC.10.228. PMID 21426138. edit
- ^ Sproule, D. M.; Kaufmann, P. (2010). "Therapeutic developments in spinal muscular atrophy". Therapeutic Advances in Neurological Disorders 3 (3): 173–185. doi:10.1177/1756285610369026. PMC 3002649. PMID 21179609. edit
- ^ Fuller, H. R.; Barišić, M.; Šešo-Šimić, Đ. I.; Špeljko, T.; Morris, G. E.; Šimić, G. (2010). "Treatment strategies for spinal muscular atrophy". Translational Neuroscience 1 (4): 308. doi:10.2478/v10134-010-0045-4. edit
- ^ Sendtner, M. (2010). "Therapy development in spinal muscular atrophy". Nature Neuroscience 13 (7): 795–799. doi:10.1038/nn.2565. PMID 20581815. edit
- ^ a b Bosboom, W. M.; Vrancken, A. F. E.; Van Den Berg, L. H.; Wokke, J. H.; Iannaccone, S. T. (2009). "Drug treatment for spinal muscular atrophy type I". In Bosboom, Wendy MJ. Cochrane Database of Systematic Reviews. doi:10.1002/14651858.CD006281.pub2. edit
- ^ Bosboom, W. M.; Vrancken, A. F. E.; Van Den Berg, L. H.; Wokke, J. H.; Iannaccone, S. T. (2009). "Drug treatment for spinal muscular atrophy types II and III". In Bosboom, Wendy MJ. Cochrane Database of Systematic Reviews. doi:10.1002/14651858.CD006282.pub2. edit
- ^ Wadman, R. I.; Bosboom, W. M.; Van Den Berg, L. H.; Wokke, J. H.; Iannaccone, S. T.; Vrancken, A. F. E. (2011). "Drug treatment for spinal muscular atrophy type I". In Wadman, Renske I. Cochrane Database of Systematic Reviews. doi:10.1002/14651858.CD006281.pub3. edit
- ^ Wadman, R. I.; Bosboom, W. M.; Van Den Berg, L. H.; Wokke, J. H.; Iannaccone, S. T.; Vrancken, A. F. E. (2011). "Drug treatment for spinal muscular atrophy types II and III". In Wadman, Renske I. Cochrane Database of Systematic Reviews. doi:10.1002/14651858.CD006282.pub3. edit
- ^ Lewelt, A.; Newcomb, T. M.; Swoboda, K. J. (2011). "New Therapeutic Approaches to Spinal Muscular Atrophy". Current Neurology and Neuroscience Reports 12 (1): 42–53. doi:10.1007/s11910-011-0240-9. PMC 3260050. PMID 22134788. edit
- ^ Passini, M. A.; Cheng, S. H. (2011). "Prospects for the gene therapy of spinal muscular atrophy". Trends in Molecular Medicine 17 (5): 259–265. doi:10.1016/j.molmed.2011.01.002. PMID 21334976. edit
- ^ Bevan, A. K.; Duque, S.; Foust, K. D.; Morales, P. R.; Braun, L.; Schmelzer, L.; Chan, C. M.; McCrate, M.; Chicoine, L. G.; Coley, B. D.; Porensky, P. N.; Kolb, S. J.; Mendell, J. R.; Burghes, A. H.; Kaspar, B. K. (2011). "Systemic Gene Delivery in Large Species for Targeting Spinal Cord, Brain, and Peripheral Tissues for Pediatric Disorders". Molecular Therapy 19 (11): 1971–1980. doi:10.1038/mt.2011.157. PMC 3222525. PMID 21811247. edit
- ^ Evans, M. C.; Cherry, J. J.; Androphy, E. J. (2011). "Differential regulation of the SMN2 gene by individual HDAC proteins". Biochemical and Biophysical Research Communications 414 (1): 25–30. doi:10.1016/j.bbrc.2011.09.011. PMID 21925145. edit
- ^ Chang, J. -G.; Hsieh-Li, H. -M.; Jong, Y. -J.; Wang, N. M.; Tsai, C. -H.; Li, H. (2001). "Treatment of spinal muscular atrophy by sodium butyrate". Proceedings of the National Academy of Sciences 98 (17): 9808. doi:10.1073/pnas.171105098. edit
- ^ Andreassi, C.; Angelozzi, C.; Tiziano, F. D.; Vitali, T.; De Vincenzi, E.; Boninsegna, A.; Villanova, M.; Bertini, E.; Pini, A.; Neri, G.; Brahe, C. (2003). "Phenylbutyrate increases SMN expression in vitro: Relevance for treatment of spinal muscular atrophy". European Journal of Human Genetics 12 (1): 59–65. doi:10.1038/sj.ejhg.5201102. PMID 14560316. edit
- ^ Brahe, C.; Vitali, T.; Tiziano, F. D.; Angelozzi, C.; Pinto, A. M.; Borgo, F.; Moscato, U.; Bertini, E.; Mercuri, E.; Neri, G. (2004). "Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients". European Journal of Human Genetics 13 (2): 256–259. doi:10.1038/sj.ejhg.5201320. PMID 15523494. edit
- ^ Mercuri, E.; Bertini, E.; Messina, S.; Solari, A.; d'Amico, A.; Angelozzi, C.; Battini, R.; Berardinelli, A.; Boffi, P.; Bruno, C.; Cini, C.; Colitto, F.; Kinali, M.; Minetti, C.; Mongini, T.; Morandi, L.; Neri, G.; Orcesi, S.; Pane, M.; Pelliccioni, M.; Pini, A.; Tiziano, F. D.; Villanova, M.; Vita, G.; Brahe, C. (2007). "Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy". Neurology 68 (1): 51–55. doi:10.1212/01.wnl.0000249142.82285.d6. PMID 17082463. edit
- ^ "Study to Evaluate Sodium Phenylbutyrate in Pre-symptomatic Infants With Spinal Muscular Atrophy (STOPSMA)". Retrieved 28 December 2011.
- ^ Brichta, L.; Hofmann, Y.; Hahnen, E.; Siebzehnrubl, F. A.; Raschke, H.; Blumcke, I.; Eyupoglu, I. Y.; Wirth, B. (2003). "Valproic acid increases the SMN2 protein level: A well-known drug as a potential therapy for spinal muscular atrophy". Human Molecular Genetics 12 (19): 2481–2489. doi:10.1093/hmg/ddg256. PMID 12915451. edit
- ^ Tsai, L. K.; Tsai, M. S.; Ting, C. H.; Li, H. (2008). "Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice". Journal of Molecular Medicine 86 (11): 1243–1254. doi:10.1007/s00109-008-0388-1. PMID 18649067. edit
- ^ Swoboda, K. J.; Scott, C. B.; Crawford, T. O.; Simard, L. R.; Reyna, S. P.; Krosschell, K. J.; Acsadi, G.; Elsheik, B.; Schroth, M. K.; d'Anjou, G.; Lasalle, B.; Prior, T. W.; Sorenson, S. L.; MacZulski, J. A.; Bromberg, M. B.; Chan, G. M.; Kissel, J. T.; Project Cure Spinal Muscular Atrophy Investigators Network (2010). "SMA CARNI-VAL Trial Part I: Double-Blind, Randomized, Placebo-Controlled Trial of L-Carnitine and Valproic Acid in Spinal Muscular Atrophy". In Boutron, Isabelle. PLoS ONE 5 (8): e12140. doi:10.1371/journal.pone.0012140. PMC 2924376. PMID 20808854. edit
- ^ Kissel, J. T.; Scott, C. B.; Reyna, S. P.; Crawford, T. O.; Simard, L. R.; Krosschell, K. J.; Acsadi, G.; Elsheik, B.; Schroth, M. K.; d'Anjou, G.; Lasalle, B.; Prior, T. W.; Sorenson, S.; MacZulski, J. A.; Bromberg, M. B.; Chan, G. M.; Swoboda, K. J.; Project Cure Spinal Muscular Atrophy Investigators' Network (2011). "SMA CARNI-VAL TRIAL PART II: A Prospective, Single-Armed Trial of L-Carnitine and Valproic Acid in Ambulatory Children with Spinal Muscular Atrophy". In Feany, Mel B. PLoS ONE 6 (7): e21296. doi:10.1371/journal.pone.0021296. PMC 3130730. PMID 21754985. edit
- ^ Darbar, I. A.; Plaggert, P. G.; Resende, M. B. D.; Zanoteli, E.; Reed, U. C. (2011). "Evaluation of muscle strength and motor abilities in children with type II and III spinal muscle atrophy treated with valproic acid". BMC Neurology 11: 36. doi:10.1186/1471-2377-11-36. PMC 3078847. PMID 21435220. edit
- ^ Rak, K.; Lechner, B. D.; Schneider, C.; Drexl, H.; Sendtner, M.; Jablonka, S. (2009). "Valproic acid blocks excitability in SMA type I mouse motor neurons". Neurobiology of Disease 36 (3): 477–487. doi:10.1016/j.nbd.2009.08.014. PMID 19733665. edit
- ^ Riessland, M.; Brichta, L.; Hahnen, E.; Wirth, B. (2006). "The benzamide M344, a novel histone deacetylase inhibitor, significantly increases SMN2 RNA/protein levels in spinal muscular atrophy cells". Human Genetics 120 (1): 101–110. doi:10.1007/s00439-006-0186-1. PMID 16724231. edit
- ^ Garbes, L.; Riessland, M.; Hölker, I.; Heller, R.; Hauke, J.; Tränkle, C.; Coras, R.; Blümcke, I.; Hahnen, E.; Wirth, B. (2009). "LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate". Human Molecular Genetics 18 (19): 3645–3658. doi:10.1093/hmg/ddp313. PMID 19584083. edit
- ^ Narver, H. L.; Kong, L.; Burnett, B. G.; Choe, D. W.; Bosch-Marcé, M.; Taye, A. A.; Eckhaus, M. A.; Sumner, C. J. (2008). "Sustained improvement of spinal muscular atrophy mice treated with trichostatin a plus nutrition". Annals of Neurology 64 (4): 465–470. doi:10.1002/ana.21449. PMID 18661558. edit
- ^ Avila, A. M.; Burnett, B. G.; Taye, A. A.; Gabanella, F.; Knight, M. A.; Hartenstein, P.; Cizman, Z.; Di Prospero, N. A.; Pellizzoni, L.; Fischbeck, K. H.; Sumner, C. J. (2007). "Trichostatin a increases SMN expression and survival in a mouse model of spinal muscular atrophy". Journal of Clinical Investigation 117 (3): 659–671. doi:10.1172/JCI29562. PMC 1797603. PMID 17318264. edit
- ^ Riessland, M.; Ackermann, B.; Förster, A.; Jakubik, M.; Hauke, J.; Garbes, L.; Fritzsche, I.; Mende, Y.; Blumcke, I.; Hahnen, E.; Wirth, B. (2010). "SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy". Human Molecular Genetics 19 (8): 1492–1506. doi:10.1093/hmg/ddq023. PMID 20097677. edit
- ^ Grzeschik, S. M.; Ganta, M.; Prior, T. W.; Heavlin, W. D.; Wang, C. H. (2010). "Hydroxyurea enhances SMN2 gene expression in spinal muscular atrophy cells". Annals of Neurology 58 (2): 194–202. doi:10.1002/ana.20548. PMID 16049920. edit
- ^ Chen, T. - H.; Chang, J. - G.; Yang, Y. - H.; Mai, H. - H.; Liang, W. - C.; Wu, Y. - C.; Wang, H. - Y.; Huang, Y. - B.; Wu, S. - M.; Chen, Y. - C.; Yang, S. - N.; Jong, Y. - J. (2010). "Randomized, double-blind, placebo-controlled trial of hydroxyurea in spinal muscular atrophy". Neurology 75 (24): 2190–2197. doi:10.1212/WNL.0b013e3182020332. PMID 21172842. edit
- ^ Sakla, M. S.; Lorson, C. L. (2007). "Induction of full-length survival motor neuron by polyphenol botanical compounds". Human Genetics 122 (6): 635–643. doi:10.1007/s00439-007-0441-0. PMID 17962980. edit
- ^ Dayangaç-Erden, D.; Bora, G.; Ayhan, P.; Kocaefe, Ç.; Dalkara, S.; Yelekçi, K.; Demir, A. S.; Erdem-Yurter, H. (2009). "Histone Deacetylase Inhibition Activity and Molecular Docking of (E )-Resveratrol: Its Therapeutic Potential in Spinal Muscular Atrophy". Chemical Biology & Drug Design 73 (3): 355. doi:10.1111/j.1747-0285.2009.00781.x. edit
- ^ Farooq, F.; Molina, F. A. A.; Hadwen, J.; MacKenzie, D.; Witherspoon, L.; Osmond, M.; Holcik, M.; MacKenzie, A. (2011). "Prolactin increases SMN expression and survival in a mouse model of severe spinal muscular atrophy via the STAT5 pathway". Journal of Clinical Investigation 121 (8): 3042–3050. doi:10.1172/JCI46276. PMC 3148738. PMID 21785216. edit
- ^ Angelozzi, C.; Borgo, F.; Tiziano, F. D.; Martella, A.; Neri, G.; Brahe, C. (2007). "Salbutamol increases SMN mRNA and protein levels in spinal muscular atrophy cells". Journal of Medical Genetics 45 (1): 29–31. doi:10.1136/jmg.2007.051177. PMID 17932121. edit
- ^ Pane, M.; Staccioli, S.; Messina, S.; d'Amico, A.; Pelliccioni, M.; Mazzone, E. S.; Cuttini, M.; Alfieri, P.; Battini, R.; Main, M.; Muntoni, F.; Bertini, E.; Villanova, M.; Mercuri, E. (2008). "Daily salbutamol in young patients with SMA type II". Neuromuscular Disorders 18 (7): 536–540. doi:10.1016/j.nmd.2008.05.004. PMID 18579379. edit
- ^ Tiziano, F. D.; Lomastro, R.; Pinto, A. M.; Messina, S.; d'Amico, A.; Fiori, S.; Angelozzi, C.; Pane, M.; Mercuri, E.; Bertini, E.; Neri, G.; Brahe, C. (2010). "Salbutamol increases survival motor neuron (SMN) transcript levels in leucocytes of spinal muscular atrophy (SMA) patients: Relevance for clinical trial design". Journal of Medical Genetics 47 (12): 856–858. doi:10.1136/jmg.2010.080366. PMID 20837492. edit
- ^ Andreassi, C.; Jarecki, J.; Zhou, J.; Coovert, D. D.; Monani, U. R.; Chen, X.; Whitney, M.; Pollok, B.; Zhang, M.; Androphy, E.; Burghes, A. H. (2001). "Aclarubicin treatment restores SMN levels to cells derived from type I spinal muscular atrophy patients". Human Molecular Genetics 10 (24): 2841–2849. doi:10.1093/hmg/10.24.2841. PMID 11734549. edit
- ^ Hua, Y.; Vickers, T. A.; Okunola, H. L.; Bennett, C. F.; Krainer, A. R. (2008). "Antisense Masking of an hnRNP A1/A2 Intronic Splicing Silencer Corrects SMN2 Splicing in Transgenic Mice". The American Journal of Human Genetics 82 (4): 834. doi:10.1016/j.ajhg.2008.01.014. edit
- ^ Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M. A.; Bennett, C. F.; Krainer, A. R. (2010). "Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model". Genes & Development 24 (15): 1634. doi:10.1101/gad.1941310. edit
- ^ Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C. F.; Krainer, A. R. (2011). "Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model". Nature 478 (7367): 123–126. doi:10.1038/nature10485. PMC 3191865. PMID 21979052. edit
- ^ Butchbach, M. E. R.; Singh, J.; Thorsteinsdóttir, M.; Saieva, L.; Slominski, E.; Thurmond, J.; Andrésson, T.; Zhang, J.; Edwards, J. D.; Simard, L. R.; Pellizzoni, L.; Jarecki, J.; Burghes, A. H. M.; Gurney, M. E. (2009). "Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy". Human Molecular Genetics 19 (3): 454–467. doi:10.1093/hmg/ddp510. PMC 2798721. PMID 19897588. edit
- ^ Zhang, M. L.; Lorson, C. L.; Androphy, E. J.; Zhou, J. (2001). "An in vivo reporter system for measuring increased inclusion of exon 7 in SMN2 mRNA: Potential therapy of SMA". Gene Therapy 8 (20): 1532–1538. doi:10.1038/sj.gt.3301550. PMID 11704813. edit
- ^ Burnett, B. G.; Munoz, E.; Tandon, A.; Kwon, D. Y.; Sumner, C. J.; Fischbeck, K. H. (2008). "Regulation of SMN Protein Stability". Molecular and Cellular Biology 29 (5): 1107–1115. doi:10.1128/MCB.01262-08. PMC 2643817. PMID 19103745. edit
- ^ Mattis, V. B.; Rai, R.; Wang, J.; Chang, C. W. T.; Coady, T.; Lorson, C. L. (2006). "Novel aminoglycosides increase SMN levels in spinal muscular atrophy fibroblasts". Human Genetics 120 (4): 589–601. doi:10.1007/s00439-006-0245-7. PMID 16951947. edit
- ^ Mattis, V. B.; Fosso, M. Y.; Chang, C. W.; Lorson, C. L. (2009). "Subcutaneous administration of TC007 reduces disease severity in an animal model of SMA". BMC Neuroscience 10: 142. doi:10.1186/1471-2202-10-142. PMC 2789732. PMID 19948047. edit
- ^ Lunn, M. R.; Root, D. E.; Martino, A. M.; Flaherty, S. P.; Kelley, B. P.; Coovert, D. D.; Burghes, A. H.; Thi Man, N.; Morris, G. E.; Zhou, J.; Androphy, E. J.; Sumner, C. J.; Stockwell, B. R. (2004). "Indoprofen Upregulates the Survival Motor Neuron Protein through a Cyclooxygenase-Independent Mechanism". Chemistry & Biology 11 (11): 1489–1493. doi:10.1016/j.chembiol.2004.08.024. PMC 3160629. PMID 15555999. edit
- ^ Nizzardo, M.; Nardini, M.; Ronchi, D.; Salani, S.; Donadoni, C.; Fortunato, F.; Colciago, G.; Falcone, M.; Simone, C.; Riboldi, G.; Govoni, A.; Bresolin, N.; Comi, G. P.; Corti, S. (2011). "Beta-lactam antibiotic offers neuroprotection in a spinal muscular atrophy model by multiple mechanisms". Experimental Neurology 229 (2): 214–225. doi:10.1016/j.expneurol.2011.01.017. PMID 21295027. edit
- ^ Hedlund, E. (2011). "The protective effects of beta-lactam antibiotics in motor neuron disorders". Experimental Neurology 231 (1): 14–18. doi:10.1016/j.expneurol.2011.06.002. PMID 21693120. edit
- ^ Rose, F. F.; Mattis, V. B.; Rindt, H.; Lorson, C. L. (2009). "Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy". Human Molecular Genetics 18 (6): 997–1005. doi:10.1093/hmg/ddn426. PMC 2649020. PMID 19074460. edit
- ^ Takeuchi, Y.; Miyanomae, Y.; Komatsu, H.; Oomizono, Y.; Nishimura, A.; Okano, S.; Nishiki, T.; Sawada, T. (1994). "Efficacy of Thyrotropin-Releasing Hormone in the Treatment of Spinal Muscular Atrophy". Journal of Child Neurology 9 (3): 287–289. doi:10.1177/088307389400900313. PMID 7930408. edit
- ^ Tzeng, A. C.; Cheng, J.; Fryczynski, H.; Niranjan, V.; Stitik, T.; Sial, A.; Takeuchi, Y.; Foye, P.; Deprince, M.; Bach, J. R. (2000). "A study of thyrotropin-releasing hormone for the treatment of spinal muscular atrophy: A preliminary report". American journal of physical medicine & rehabilitation / Association of Academic Physiatrists 79 (5): 435–440. doi:10.1097/00002060-200009000-00005. PMID 10994885. edit
- ^ Kato, Z.; Okuda, M.; Okumura, Y.; Arai, T.; Teramoto, T.; Nishimura, M.; Kaneko, H.; Kondo, N. (2009). "Oral Administration of the Thyrotropin-Releasing Hormone (TRH) Analogue, Taltireline Hydrate, in Spinal Muscular Atrophy". Journal of Child Neurology 24 (8): 1010–1012. doi:10.1177/0883073809333535. PMID 19666885. edit
- ^ "Roche signs agreement with PTC Therapeutics to advance treatment for Spinal Muscular Atrophy (SMA)". Retrieved 28 December 2011.
- ^ "Rethropin: drug pipeline". Retrieved 8 May 2012.
- ^ a b Grice, S. J.; Sleigh, J. N.; Liu, J. L.; Sattelle, D. B. (2011). "Invertebrate models of spinal muscular atrophy: Insights into mechanisms and potential therapeutics". BioEssays 33 (12): 956–965. doi:10.1002/bies.201100082. PMID 22009672. edit
- ^ Sleigh, J. N.; Buckingham, S. D.; Esmaeili, B.; Viswanathan, M.; Cuppen, E.; Westlund, B. M.; Sattelle, D. B. (2010). "A novel Caenorhabditis elegans allele, smn-1(cb131), mimicking a mild form of spinal muscular atrophy, provides a convenient drug screening platform highlighting new and pre-approved compounds". Human Molecular Genetics 20 (2): 245–260. doi:10.1093/hmg/ddq459. PMID 20962036. edit
- ^ Chang, H. C. H.; Dimlich, D. N.; Yokokura, T.; Mukherjee, A.; Kankel, M. W.; Sen, A.; Sridhar, V.; Fulga, T. A.; Hart, A. C.; Van Vactor, D.; Artavanis-Tsakonas, S. (2008). "Modeling Spinal Muscular Atrophy in Drosophila". In Lewin, Alfred. PLoS ONE 3 (9): e3209. doi:10.1371/journal.pone.0003209. PMC 2527655. PMID 18791638. edit
- ^ Grice, S. J.; Liu, J. L. (2011). "Survival Motor Neuron Protein Regulates Stem Cell Division, Proliferation, and Differentiation in Drosophila". In Rulifson, Eric. PLoS Genetics 7 (4): e1002030. doi:10.1371/journal.pgen.1002030. PMC 3072375. PMID 21490958. edit
- ^ Beattie, C. E.; Carrel, T. L.; McWhorter, M. L. (2007). "Fishing for a Mechanism: Using Zebrafish to Understand Spinal Muscular Atrophy". Journal of Child Neurology 22 (8): 995–1003. doi:10.1177/0883073807305671. PMID 17761655. edit
- ^ Sleigh, J. N.; Gillingwater, T. H.; Talbot, K. (2011). "The contribution of mouse models to understanding the pathogenesis of spinal muscular atrophy". Disease Models & Mechanisms 4 (4): 457–467. doi:10.1242/dmm.007245. PMC 3124050. PMID 21708901. edit
- ^ "The GSF and FightSMA Announce 100K Research Award for the Development of a Large Animal Model of Spinal Muscular Atrophy". Retrieved 18 December 2011.
- ^ Le, T. T.; McGovern, V. L.; Alwine, I. E.; Wang, X.; Massoni-Laporte, A.; Rich, M. M.; Burghes, A. H. M. (2011). "Temporal requirement for high SMN expression in SMA mice". Human Molecular Genetics 20 (18): 3578–3591. doi:10.1093/hmg/ddr275. PMC 3159555. PMID 21672919. edit
- ^ Porensky, P. N.; Mitrpant, C.; McGovern, V. L.; Bevan, A. K.; Foust, K. D.; Kaspar, B. K.; Wilton, S. D.; Burghes, A. H. M. (2011). "A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in the mouse". Human Molecular Genetics 21 (7): 1625–38. doi:10.1093/hmg/ddr600. PMID 22186025. edit
94. Parano E; Pavone L, Falsaperla R, Trifiletti R, Wang C (1996). Molecular basis of phenotypic heterogeneity in sibling with Spinal Muscular Atrophy. Annals of Neurology (40): 247-251.
External links[edit]
- SMA at NINDS
- Spinal muscular atrophy on the Open Directory Project
- Standards of Care in Spinal Muscular Atrophy
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||
UpToDate Contents
全文を閲覧するには購読必要です。 To read the full text you will need to subscribe.
- 1. 脊髄性筋萎縮症 spinal muscular atrophy
- 2. 肢帯型筋ジストロフィー limb girdle muscular dystrophy
- 3. 神経筋疾患の評価における筋酵素 muscle enzymes in the evaluation of neuromuscular diseases
English Journal
- Spinal muscular atrophy due to double gene conversion event.
- Maamouri W, Hammer MB, Bouhlel Y, Souilem S, Khmiri N, Nehdi H, Hentati F, Amouri R.SourceDepartment of Molecular Neurobiology and Neuropathology, National Institute of Neurology, La Rabta, Tunis, Tunisia. wieme hicheri@yahoo.fr
- The International journal of neuroscience.2011 Feb;121(2):107-11. Epub 2010 Nov 3.
- Spinal muscular atrophy (SMA) is an autosomal recessive disease characterized by degeneration of the anterior horn cells of the spinal cord. The survival motor neuron (SMN) gene has been identified as an SMA-determining gene. SMN exists as two copies in 5q13, and deletions in exons 7 and 8 of the te
- PMID 21047176
- Altered axonal excitability properties in juvenile muscular atrophy of distal upper extremity (Hirayama disease).
- Sawai S, Misawa S, Kanai K, Isose S, Shibuya K, Noto Y, Fujimaki Y, Sekiguchi Y, Nasu S, Nomura F, Kuwabara S.SourceDepartment of Molecular Diagnosis, Graduate School of Medicine, Chiba University, 1-8-1 Inohana, Chuo-ku, Chiba 260-8670, Japan. ssawai@yj8.so-net.ne.jp
- Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology.2011 Jan;122(1):205-9. Epub 2010 Jul 10.
- PMID 20624687
Japanese Journal
- An autopsy case of spinal muscular atrophy type III (Kugelberg-Welander disease)
- KURU Satoshi,SAKAI Motoko,KONAGAYA Masaaki,YOSHIDA Mari,HASHIZUME Yoshio,SAITO Kayoko
- Neuropathology : official journal the Japanese Society of Neuropathology 29(1), 63-67, 2009-02-01
- NAID 10025824921
- Kugelberg-Welander病に合併した自然気胸の1手術例
- 近藤 正道,南 寛行,原 信介,宮崎 拓郎
- 日本呼吸器外科学会雑誌 19(1), 56-59, 2005-01-15
- … 症例は46歳,男性.16歳時に遺伝性若年性近位性脊髄性筋萎縮症(Kugelberg-Welander病)を発症し,入院時は四肢挙上不能の状態であった.2003年2月に夜間に咳嗽が出現.当院における胸部X線写真にて右気胸と診断され入院となった.保存的治療を試みるも治癒せず,3月に全身麻酔下に胸腔鏡補助下小開胸併用にて肺部分切除を施行した.筋弛緩には臭化ベクロニウムを用いたが,術直後の回復は軽度遅延した.また術後3 …
- NAID 10014315726
Related Links
- 世界大百科事典 第2版 Kugelberg-Welander diseaseの用語解説 - 急速に進行して四肢麻痺を生じ,数年で死亡する。クーゲルベルク=ウェランダー病Kugelberg‐Welander diseaseは,主として肩甲部や腰部など四肢の体幹に近い部位の筋肉 ...
- Kugelberg-Welander disease SNOMED CT Kugelberg-Welander disease, ID: 54280009 Synonyms Juvenile Spinal Muscular Atrophy, Kugelberg-Welander syndrome, Muscular atrophy, juvenile, SMA 3, SMA III, SPINAL MUSCULAR ...
Related Pictures


.jpg)

★リンクテーブル★
| リンク元 | 「脊髄性筋萎縮症」「脊髄性筋萎縮症3型」「運動ニューロン疾患」「神経変性疾患」 |
| 関連記事 | 「disease」 |
「脊髄性筋萎縮症」
- 英
- spinal muscular atrophy, SMA
- 同?
- 脊髄進行性筋萎縮症 progressive spinal muscular atrophy
- 関
- 筋萎縮、運動ニューロン疾患, motor neuron disease, MND
運動ニューロン病
- BET.439
- 上位運動ニューロン and/or 下位運動ニューロンが選択的に傷害される疾患の総称
| 上位運動ニューロン障害 | 原発性側索硬化症 PLS | ||
| 上位+下位運動ニューロン障害 | 筋萎縮性側索硬化症 ALS | 孤発性/家族性 | |
| 下位運動ニューロン障害 | 脊髄性筋萎縮症 SMA | SMA type 1, infantile SMA, Werdnig-Hoffmann disease ウェルドニッヒ・ホフマン病 WH | AR |
| SMA type 2, intermediate SMA | |||
| SMA type 3, juvenile SMA, Kugelberg-Welander disease クーゲルベルク・ウェランダー病 KW | AR | ||
| 球脊髄性筋萎縮症 BSMA Kennedy-Alter-Sung disease ケネディ・オルター・スン症候群 KAS | XR、CAGリピート | ||
遺伝形式
原因遺伝子
- 5q13.2に座乗するSMN1遺伝子の変異。
症状
- 下位運動ニューロンの脱落・変性
- 近位筋優位の筋萎縮 ← 神経原性の筋萎縮なのに・・・ (cf. 筋萎縮)
参考
uptodate
- 1. [charged] 脊髄性筋萎縮症 - uptodate [1]
OMIM
- 1. SPINAL MUSCULAR ATROPHY, TYPE I; SMA1, Gene map locus 5q12.2-q13.3
- 2. SPINAL MUSCULAR ATROPHY, TYPE II; SMA2, Gene map locus 5q12.2-q13.3
- 3. SPINAL MUSCULAR ATROPHY, TYPE III; SMA3, Gene map locus 5q12.2-q13.3, 5q12.2-q13.3
- 4. SPINAL MUSCULAR ATROPHY, TYPE IV; SMA4, Gene map locus 5q12.2-q13.3
「脊髄性筋萎縮症3型」
- 英
- spinal muscular atrophy type 3 SMA 3
- 同
- クーゲルベルク・ウェランダー病 クーゲルバーグ・ウェランダー病 クーゲルベルク-ヴェランデル病 クーゲルベルク-ヴェランダー病 クーゲルベルク・ヴェランダー病 Kugelberg-Welander病 Kugelberg-Welander disease KW disease K-W disease, ヴォールファルト-クーゲルベルク-ヴェランダー病 Wohlfart-Kugelberg-Welander disease、若年性進行性脊髄性筋萎縮症 juvenile progressive spinal muscular atrophy
- 関
- 脊髄性筋萎縮症 SMA
クーゲルベルク・ウェランダー病 : 46 件 クーゲルベルク・ヴェランダー病 : 39 件 クーゲルバーグ・ウェランダー病 : 約 16 件 クーゲルベルク・ヴェランデル病 : 約 9 件
「運動ニューロン疾患」
- 英
- motor neuron disease, MND
- 関
- 筋萎縮
- 神経系 091102IV
運動ニューロン病(BET.439)
- 上位運動ニューロン and/or 下位運動ニューロンが選択的に傷害される疾患の総称
| 上位運動ニューロン障害 | 原発性側索硬化症 PLS | |
| 上位+下位運動ニューロン障害 | 筋萎縮性側索硬化症 ALS | |
| 下位運動ニューロン障害 | 脊髄性筋萎縮症 SMA | SMA type 1, infantile SMA, Werdnig-Hoffmann disease |
| SMA type 2, intermediate SMA | ||
| SMA type 3, juvenile SMA, Kugelberg-Welander disease | ||
| 球脊髄性筋萎縮症 BSMA Kennedy-Alter-Sung disease | ||
「神経変性疾患」
- 英
- neurodegenerative disease, neurodegenerative diseases, neural degenerative disorder, degenerative cerebellar disease, degenerative neurologic disease, neurologic degenerative disease, nervous system degenerative disease
- 関
- 中枢神経変性疾患
- I型 Werding-Hoffmann disease ウエルドニッヒ・ホフマン病
- II型
- III型 Kugelberg-Welander disease クーゲルベルク・ウェランダー病
- 球脊髄性筋萎縮症 spinal bulbar muscular atrophy SBMA Kennedy-Alter-Sung disease ケネディ・オルター・スン症候群
「disease」
- n.
- 疾患:illnessより厳密な概念。「ある臓器に明確な障害が確認され、それによって症状が出ているとはっきり説明できる場合」 (PSY.9)
- 特定の原因、病態生理、症状、経過、予後、病理組織所見が全てそろった場合 (PSY.9)
- something that is very wrong with people's attitudes, way of life or with society.
- 注意