|
|
この記事は検証可能な参考文献や出典が全く示されていないか、不十分です。
出典を追加して記事の信頼性向上にご協力ください。(2011年10月) |
反応速度(はんのうそくど、英語: reaction velocity, reaction rate)とは化学反応の反応物あるいは生成物に関する各成分量の時間変化率を表す物理量。通常、反応速度を表現する式は濃度のべき関数として表現される。
目次
- 1 反応速度の一般式
- 2 反応速度式
- 2.1 速度定数と反応次数
- 2.2 積分形速度式
- 2.2.1 1次反応
- 2.2.2 2次反応
- 2.2.2.1 反応物が1種類の場合
- 2.2.2.2 反応物が2種類の場合
- 2.2.3 零次反応
- 3 可逆反応
- 4 単純反応と複合反応
- 5 測定方法
- 6 出典
- 7 参考文献
- 8 関連項目
反応速度の一般式
倍数比例の法則が示すように、化学反応に関与する各成分の変化量は、その間に一定の比が成り立つ従属変数であるので、特定の成分量ではなく次のような反応進行度(はんのうしんこうど)または反応進度(はんのうしんど、英: extent of reaction)ξ を定義し、その時間微分で化学反応全体の進行速度を表す。
次の一般化反応式を考える:
- 化学量数(かがくりょうすう)または化学量論係数(かがくりょうろんけいすう、英: stoichiometric number)ν(ニュー)は生成系(右辺)で正、原系または反応系(左辺)で負、すなわち
-
-
- 例えば、N2 + 3H2 → 2NH3 という化学反応では、νN2 =-1、νH2 = -3、νNH3 = 2 である。
各成分の時刻 t における物質量を n <成分>,<時刻> で表すと、反応進行度 ξ は次の各成分の物質量の時間変化の式で示すことができる。
したがって反応速度 v (または、rateの頭文字をとってr )は、反応進行度あるいは各成分の物質量の時間変化で、次のように定義される。
物質量nA と容積V およびモル濃度cA との関係は
で表される。したがって化学反応が時間変化しない一定の容積内で進行する場合には、前述の反応速度v は物質のモル濃度変化vc で表すことができる。一般に、このvc のことをv と書くことが多い。
ところで一般に反応系が平衡から大きく外れている場合反応速度は濃度のべき関数として近似可能なので反応速度を反応物濃度を使って次の式で表現する。
一般に反応速度を表すべき関数のべき乗係数の総和n を全反応次数(ぜんはんのうじすう、overall reaction order)と呼び反応速度式を分類する目的で利用される。また係数k はn 次の速度定数(そくどていすう、rate constant)と呼ぶ。なお、べき乗係数p, q , ... と化学量数 νA, νB, ... との間には直接の関係はない。
反応速度式
速度定数と反応次数
一般に反応が進行中のとき、計測された任意の時間t における反応速度は、濃度の累乗に比例した値に近似できる。ゆえに反応速度は反応物濃度を使って次の式で表すことができる[1]。
右辺のような式を反応速度式(rate law)という。またある化学種(反応に関わるそれぞれの化学物質)についたべき数をその化学種に対する反応の次数(order)と呼ぶ。例えばv =kr[A][B]2 で表される速度式を持つ反応ではAについて1次、Bについて2次である。次数は整数であるとは限らず、多くの気相反応では0.5など整数ではない次数をとる[2]。反応速度を表すべき関数のべき乗係数の総和n を全反応次数(ぜんはんのうじすう、overall reaction order)と呼び[2]反応速度式を分類する目的で利用される。また係数k はn 次の速度定数(そくどていすう、rate constant)と呼ぶ[3]。速度定数は反応物および生成物の濃度には依存せず、系の温度のみに依存する定数である[4]。なお、反応係数p, q, ... と化学量数νA, νB, ... との間には直接の関係はない。反応次数は経験的にわかる濃度依存性を表している[4]。
積分形速度式
速度式は微分方程式である。速度式を積分することで時間に対する濃度の関数を得ることができる[5]。
1次反応
においてAの初濃度が[A]0のとき、時間t の後、x mol/dm³ (= [B]) が反応したとする 。すると[A]0 - x は時間t におけるAの濃度[A]に等しくなる。Bの生成速度dx /dt は[A]に比例するから、反応速度定数をk1 とすると
または
という式で表すことができる[5]。この微分方程式を
のように変形し、両辺をそれぞれ[A]0から[A]、0からt で積分すると、
と書くことができる[6]。1/[A]の積分はln [A]であることから次の積分系速度式が得られる[7]。
生成物Bの濃度に対しては
という式が得られる。
1次反応では反応物は初期濃度から指数関数的に減少する。その速度は速度定数k1 のみで決定される。場合によっては速度定数の代わりに半減期で速度を表す場合もある。半減期と1次の速度定数と間には次の関係がある。
2次反応
化学反応が2次反応であるとき、2つの型が考えられる。反応物が1種類である場合と異なる2つの物質が反応に関与する場合である[8]。
反応物が1種類の場合
2次反応で、反応物が1種類の時の反応は一般的に次のようなものである[8]。
反応物Aの初濃度を[A]0とし、t 時間反応したとすると、反応速度式は
と表すことができる[9]。変数[A], t を分離してこの式を積分すると
と書くことができる[9]。計算は以下のボックス中に示す。
計算[9][8]
2次反応の速度式を
と変形する。両辺をそれぞれ[A]0から[A]、0からt の範囲で積分すると、
となる。1/x2の積分は-1/x であることから、次の積分系速度式が得られる。
より、1/[A]をt に対してプロットすると、傾きk2 、切片1/[A]0 の直線が得られる[8]。
反応物が2種類の場合
反応物が2種類の2次反応は、次のような式になる。
この時Aが1次、Bが1次で合計2次の反応になる[8]。反応速度定数をk2 として、時間t におけるAの濃度[A]またはBの濃度[B]の反応速度式をたてると
となる[8]。Aの初濃度を[A]0、Bの初濃度を[B]0とし、時間tののちx mol/dm³が反応したとする。すると生成物Cの生成速度dx /dt は[A]および[B]に比例する。また[A] = [A]0 - x 、[B] = [B]0 - x であるから
となり、生成物Cの生成速度式は
となる[10]。この式に部分積分法を用いて積分すると、最終的には以下のような式が得られる[11][10]。
計算方法は以下のボックスに示す。
計算[10]
生成物Cの生成速度式を以下のように変形する
t = 0のときx = 0であることを用いて積分を行うと
となる。右辺の積分は単純にk2 t と導くことができる。左辺の積分は部分積分法を使う。まず
の置き換えを用いると
となるので、a, b に[A]0, [B]0を代入すると
となる。[A] = [A]0 - x、[B] = [B]0 - x であり、さらにlny - lnz = ln(y /z ) より2つの対数をまとめると次の積分系速度式が得られる。
2次反応では半減期は各時間の濃度に反比例して長くなる。初期濃度a の半分の濃度になる時間t50(すなわち、反応が50%まで進行する所要時間)は次の初期濃度a の関数で表される。
成分a とb との初期濃度が著しく相違し、の場合、2次速度式の微分方程式はさらに成分a の1次速度式に近似することができる。この場合の成分a の1次速度式の速度定数は擬1次速度定数(ぎいちじそくどていすう、pseudo-first order rate constant)と呼ばれる。
零次反応
反応が反応系の成分濃度や分圧に無関係に進行する場合は、反応速度式の全反応次数は0となり零次反応(れいじはんのう、zero-order reaction)と呼ばれる。たとえば触媒反応において触媒表面に大量の反応物が吸着して飽和状態になっており触媒への吸着過程が律速段階になっていない等、特別の環境下での反応においては当該成分濃度項の反応次数は0として近似され
の式で表すことができる[4]。k0 が零次反応の速度定数である。この式から零次反応の速度は反応物の濃度に依存しないことがわかる。またこの式を0からt ([A]0から[A]) の範囲で積分すると
となり、濃度[A]は時間t に依存することがわかる[4]。
可逆反応
前節で示した反応速度式はすべて生成物から反応物に戻る反応 (逆反応) を無視している。しかし多くの反応はある程度可逆的であり、逆反応も考慮しなくてはならない。特に反応が平衡に近づいた時は系の中に反応物が大量に存在しているので、逆反応が無視できなくなる[12]。 AからBが生成する反応で、正反応と逆反応の両方が1次のとき、次のような反応様式となる。
- A→B 反応速度 = k1[A]
- B→A 反応速度 = k-1[B]
正反応によってAの濃度[A]がk1[A]の速度で減少し、逆反応によってk-1[B]の速度で増加する。したがって[A]の正味の変化速度は
である[12]。t →∞で反応が平衡状態になるとAの正味の濃度変化は無くなるので、 であり
となる[13]。[A]eqは平衡状態でのAの濃度、[B]eqは平衡状態でのBの濃度である。この式から
を導くことができ、このK を平衡定数と呼ぶ[13]。この式は熱力学的な量である平衡定数と反応速度に関わる量である速度定数の関係を表す重要な式である。平衡定数と片方の速度定数が明らかであれば、計算によりもう片方の速度定数を求めることができる[12]。
単純反応と複合反応
反応速度の全反応次数は反応の原系の成分数と合致することが反応速度式の解釈から期待されるが、実際の反応では成分数よりも少ない反応次数の速度となることが多い。その原因の多くは目的の反応が反応式で書き表されている反応物から生成物が直接生成する単純反応(たんじゅんはんのう、simple reaction)ではなく、反応式には現れない反応中間体(はんのうちゅうかんたい、reaction intermediate, intermediate product)を介した複数の反応過程を経由する複合反応(ふくごうはんのう、complex reaction)であることによる。反応中間体は単に中間体と呼ばれることもある。
反応を考えるとき、物質変化する1つの過程を素反応(elementary reaction)と呼ぶ[14]。この場合で、物質変化が物理変化の場合は、反応素過程(elementary process of reaction)と呼ばれ、反応中間体に相当する物理状態が遷移状態である。反応素過程も含んで素反応と言い表す場合もある。この反応で、反応物の物質の数を反応分子数(molecularity)という。たとえば、以下の反応の反応分子数は2である。
H2+Cl2→2HCl
言い換えると、単純反応の場合は単一の素反応で構成されるが、複合反応は複数の素反応と反応中間体を含んで反応が構成されることになる。素反応を介して反応物から反応中間体を経て生成物に至るので、複合反応は連続反応(れんぞくはんのう、successive reaction, consecutive reaction)、逐次反応(ちくじはんのう、consecutive reaction)、連鎖反応(れんさはんのう、chain reaction)とも呼ばれる。
ある反応中間体(あるいは反応物)から2つの素反応が分岐する場合の連続反応は平行反応(parallel reaction)と呼ばれる。平行反応はラジカル反応等ではしばしば見られる素反応構成である。
複合反応を構成する素反応のそれぞれの反応速度が同一であることは少なく、(道路の自然渋滞の先頭車両を見出すことができないことと同様で)反応進行度の変化点である反応中間体は反応系内に存在するものの観測しにくいことが多い。それ故、反応中間体の存在は直接観測されるのではなかった。 反応中間体は、各種の分光法による直接観測や立体障害などで後続の反応を妨害することによる安定化、反応中間体と選択的に反応する試薬によるトラップなどの方法を使い、反応速度や反応機構からその存在が推定される場合が多かった。しかし近年は、分析技術の向上により反応中間体を直接観測できるようになりつつあり、または計算機実験による反応経路の評価などによって存在が推定されている。
逐次反応の速度式
逐次反応も単純反応と同様に時間と濃度の微分方程式をたてることで、任意の時間の反応速度や成分濃度を求めることができる。逐次反応は次のように素反応過程の生成物が次の過程の反応物となる[14][15]。
各反応が一次反応であるなら、Aの減少速度は
である[14]。BはAからk1[A]の速度で生成される一方k2[B]の速度でCに変換されるため、正味の生成速度は
であり、Cの生成速度は
である[16]。始めAのみが存在していたとし、その時の濃度を[A]0とすると、一次反応の積分系速度式より
と表すことができる。(2) を (1b) に代入して整理すると
となり、この微分方程式を解くと
となる[16]。 であるので
である[16]。
反応が2ステップを超えるとたちまち反応速度式は複雑になってしまう。しかし定常状態の近似 (steady‐state approximation) (→定常状態)という手法を用いることで数学的な処理を簡単にすることができる。これは反応初期の誘導期間のあと、主要な反応が起こっている間中間体 (今回の例ではB) の濃度はほぼ一定であると仮定する手法である。つまり
とおくことができる[15]。
律速段階
逐次反応において最も遅い素反応(過程)を律速段階(りっそくだんかい、rate-determining step)と呼ぶ。あるいは律速過程とも言う。それは最も遅い素反応(過程)が、複合反応の反応速度に対して強い影響を及ぼし、その反応の振る舞いを決定づける為である。
測定方法
前述の定義のように、反応速度を決定するには物質変化を定量分析することで測定する。反応速度がかなり遅い場合は反応系をサンプリングして容量分析することも可能であるが、大抵の場合は測定時間が短い分光法分析による定量分析が必要になる。反応速度が早い場合は反応装置や反応系にも工夫が施される。近年では高速のレーザーパルスを利用することでフェムト秒やアト秒の物質状態を分光測定することも可能になり極めて高速の反応過程も観測できる。
高速流通法
高速流通法(こうそくりゅうつうほう、rapid-flow method)では反応器とそこから引き出された管路の先に固定された分光定量装置を用意する。反応器にシリンジで反応成分を注入混合されてスタートした反応液は、引き続き管路から流出させる。そのことにより測定器の前を連続的に反応液が通過するので成分の経時変化が測定できる。連続フロー法 (continuous flow method) とも呼ばれる。高速流通法では大量の反応液が必要なため、反応液の通過を止めて測定する場合はストップトフロー法 (stopped flow method) と呼ばれ、種々のプローブを使ういくつかの方法が開発されている。特に円偏光二色性を利用する場合には蛋白質の2次構造の変化を、X線溶液散乱法と結合されたときには蛋白質のコンパクトネスを観測するのに有効である。
緩和法
また、平衡状態にある反応に対して反応系の温度や圧力等を変化させ、新たな条件での平衡点へと化学反応が進行する過程を解析する反応速度の測定方法を緩和法(かんわほう、relaxation method)と呼ぶ。温度変化を利用する場合は温度ジャンプ法(おんど—ほう、temperature jump)、圧力変化を利用する場合は圧力ジャンプ法(あつりょく—ほう、pressure jump method)と呼ばれる。
レーザーを使って温度を上げる装置を用いる場合はレーザー温度ジャンプ法という。これは非常に短時間(およそ10ナノ秒程度)で温度を上げることができるので、速い反応の解析に用いられる。特に最近では蛋白質のフォールディングの初期反応の解析に用いられて大きな成果をあげている。
出典
- ^ Atkins et al.,2011 p.658-659
- ^ a b Atkins et al.,2011 p.659
- ^ Atkins et al. ,2011 p.658
- ^ a b c d Chang p.188
- ^ a b Atkins et al.,2011 p.660
- ^ Atkins et al.,2011 p.660-661
- ^ Atkins et al.,2011 p.661
- ^ a b c d e f Chang, 2006 p.190
- ^ a b c Atkins et al.,2011 p.662
- ^ a b c Atkins et al.,2011 p.663
- ^ Chang, 2006 p.191
- ^ a b c Atkins et al.,2011 p.664
- ^ a b Chang, 2006 p.196
- ^ a b c Atkins et al.,2011 p.667
- ^ a b Chang, 2006 p.197
- ^ a b c Atkins et al.,2011 p.668
参考文献
- Peter Atkins・Julio de Paula・Ronald Friedman 『アトキンス基礎物理化学(下)―分子論的アプローチ―』 千原 秀昭・稲葉 章 訳、東京化学同人、2011年。ISBN 9784807907519。
- Raymond Chang 『生命科学系のための物理化学』 岩澤康裕・濱口 宏夫・北川 禎三 訳、東京化学同人、2006年。ISBN 9784807906451。
関連項目
- 反応速度論
- アレニウスの式
- ミカエリス・メンテン式
- 化学平衡
- 触媒
- 遷移状態
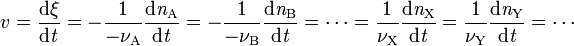

![v_\mathrm{c} = \frac{1}{V}\frac{\mathrm{d}\xi}{\mathrm{d}\mathit{t}}
= -\frac{1}{-\nu_\mathrm{A}}\frac{\mathrm{d}[\mathrm{A}]}{\mathrm{d}t}
= k[\mathrm{A}]^p [\mathrm{B}]^q [\mathrm{C}]^r \cdots](http://upload.wikimedia.org/math/0/9/3/0939da9c70f799a364cbce89a48aa7d5.png)
![v_\mathrm{c}
= \frac{1}{V}\frac{\mathrm{d}\xi}{\mathrm{d}t}
= -\frac{1}{-\nu_\mathrm{A}}\frac{\mathrm{d}[\mathrm{A}]}{\mathrm{d}t}
= k[\mathrm{A}]^p [\mathrm{B}]^q [\mathrm{C}]^r \cdots](http://upload.wikimedia.org/math/d/d/f/ddfe8cd13c8deba41ea0c64e6c64123e.png)

![\begin{align}
&\int_{0}^{x}{\frac{\mathrm{d}x}{([\mathrm{A}]_0-x)([\mathrm{B}]_0-x)}}\\
&=\frac{1}{[\mathrm{B}]_0-[\mathrm{A}]_0}\left(\ln\frac{1}{[\mathrm{A}]_0-x}-\ln\frac{1}{[\mathrm{B}]_0-x}\right)
-\frac{1}{[\mathrm{B}]_0-[\mathrm{A}]_0}(\ln[\mathrm{B}]_0-\ln[\mathrm{A}]_0)\\
&=\frac{1}{[\mathrm{B}]_0-[\mathrm{A}]_0}\left(\ln\frac{[\mathrm{A}]_0}{[\mathrm{A}]_0-x}-\ln\frac{[\mathrm{B}]_0}{[\mathrm{B}]_0-x}\right)
\end{align}](http://upload.wikimedia.org/math/d/a/2/da2537b938c87ec0827744f36e070544.png)






