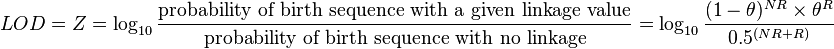"Genetic map" redirects here. It is not to be confused with Gene map.
Genetic linkage is the tendency of genes that are located proximal to each other on a chromosome to be inherited together during meiosis. Genes whose loci are nearer to each other are less likely to be separated onto different chromatids during chromosomal crossover, and are therefore said to be genetically linked.
|
Contents
- 1 Discovery
- 2 Linkage map
- 3 LOD score method for estimating recombination frequency
- 4 Recombination frequency
- 5 Meiosis Indicators
- 6 See also
- 7 References
- 8 External links
|
Discovery
Genetic linkage was first discovered by the British geneticists William Bateson and Reginald Punnett shortly after Mendel's laws were rediscovered. The understanding of genetic linkage was expanded by the work of Thomas Hunt Morgan. Morgan's observation that the amount of crossing over between linked genes differs led to the idea that crossover frequency might indicate the distance separating genes on the chromosome.
Alfred Sturtevant, a student of Morgan's, first developed genetic maps, also known as linkage maps. Sturtevant proposed that the greater the distance between linked genes, the greater the chance that non-sister chromatids would cross over in the region between the genes. By working out the number of recombinants it is possible to obtain a measure for the distance between the genes. This distance is expressed in terms of a genetic map unit (m.u.), or a centimorgan and is defined as the distance between genes for which one product of meiosis in 100 is recombinant. A recombinant frequency (RF) of 1% is equivalent to 1 m.u. But this equivalence is only a good approximation for small percentages; the largest percentage of recombinants cannot exceed 50%, which would be the situation where the two genes are at the extreme opposite ends of the same chromosomes. In this situation, any crossover events would result in an exchange of genes, but only an odd number of crossover events (a 50-50 chance between even and odd number of crossover events) would result in a recombinant product of meiotic crossover. A statistical interpretation of this is through the Haldane mapping function[1] [2] or the Kosambi mapping function,[3] among others. A linkage map is created by finding the map distances between a number of traits that are present on the same chromosome, ideally avoiding having significant gaps between traits to avoid the inaccuracies that will occur due to the possibility of multiple recombination events.
Linkage map
A linkage map is a genetic map of a species or experimental population that shows the position of its known genes or genetic markers relative to each other in terms of recombination frequency, rather than a specific physical distance along each chromosome. Linkage mapping is critical for identifying the location of genes that cause genetic diseases.
A genetic map is a map based on the frequencies of recombination between markers during crossover of homologous chromosomes. The greater the frequency of recombination (segregation) between two genetic markers, the farther apart they are assumed to be. Conversely, the lower the frequency of recombination between the markers, the smaller the physical distance between them. Historically, the markers originally used were detectable phenotypes (enzyme production, eye color) derived from coding DNA sequences; eventually, confirmed or assumed noncoding DNA sequences such as microsatellites or those generating restriction fragment length polymorphisms (RFLPs) have been used.
Genetic maps help researchers to locate other markers, such as other genes by testing for genetic linkage of the already known markers.
A genetic map is not a physical map (such as a radiation reduced hybrid map) or gene map.
LOD score method for estimating recombination frequency
The LOD score (logarithm (base 10) of odds), developed by Newton E. Morton,[4] is a statistical test often used for linkage analysis in human, animal, and plant populations. The LOD score compares the likelihood of obtaining the test data if the two loci are indeed linked, to the likelihood of observing the same data purely by chance. Positive LOD scores favor the presence of linkage, whereas negative LOD scores indicate that linkage is less likely. Computerized LOD score analysis is a simple way to analyze complex family pedigrees in order to determine the linkage between Mendelian traits (or between a trait and a marker, or two markers).
The method is described in greater detail by Strachan and Read [1]. Briefly, it works as follows:
- Establish a pedigree
- Make a number of estimates of recombination frequency
- Calculate a LOD score for each estimate
- The estimate with the highest LOD score will be considered the best estimate
The LOD score is calculated as follows:
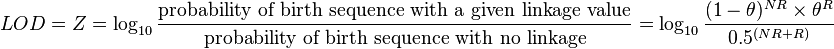
NR denotes the number of non-recombinant offspring, and R denotes the number of recombinant offspring. The reason 0.5 is used in the denominator is that any alleles that are completely unlinked (e.g. alleles on separate chromosomes) have a 50% chance of recombination, due to independent assortment.
Theta is the recombinant fraction, it is equal to R / (NR + R)
By convention, a LOD score greater than 3.0 is considered evidence for linkage. A LOD score of +3 indicates 1000 to 1 odds that the linkage being observed did not occur by chance. On the other hand, a LOD score less than -2.0 is considered evidence to exclude linkage. Although it is very unlikely that a LOD score of 3 would be obtained from a single pedigree, the mathematical properties of the test allow data from a number of pedigrees to be combined by summing the LOD scores. It is important to keep in mind that this traditional cutoff of LOD>+3 is an arbitrary one and that the difference between certain types of linkage studies, particularly analyses of complex genetic traits with hundreds of markers, these criteria should probably be modified to a somewhat higher cutoff.
Recombination frequency
Recombination frequency is a measure of genetic linkage and is used in the creation of a genetic linkage map. Recombination frequency (θ) is the frequency with which a single chromosomal crossover will take place between two genes during meiosis. A centimorgan (cM) is a unit that describes a recombination frequency of 1%. In this way we can measure the genetic distance between two loci, based upon their recombination frequency. This is a good estimate of the real distance. Double crossovers would turn into no recombination. In this case we cannot tell if crossovers took place. If the loci we're analysing are very close (less than 7 cM) a double crossover is very unlikely. When distances become higher, the likelihood of a double crossover increases. As the likelihood of a double crossover increases we systematically underestimate the genetic distance between two loci.
During meiosis, chromosomes assort randomly into gametes, such that the segregation of alleles of one gene is independent of alleles of another gene. This is stated in Mendel's Second Law and is known as the law of independent assortment. The law of independent assortment always holds true for genes that are located on different chromosomes, but for genes that are on the same chromosome, it does not always hold true.
As an example of independent assortment, consider the crossing of the pure-bred homozygote parental strain with genotype AABB with a different pure-bred strain with genotype aabb. A and a and B and b represent the alleles of genes A and B. Crossing these homozygous parental strains will result in F1 generation offspring with genotype AaBb. The F1 offspring AaBb produces gametes that are AB, Ab, aB, and ab with equal frequencies (25%) because the alleles of gene A assort independently of the alleles for gene B during meiosis. Note that 2 of the 4 gametes (50%)—Ab and aB—were not present in the parental generation. These gametes represent recombinant gametes. Recombinant gametes are those gametes that differ from both of the haploid gametes that made up the original diploid cell. In this example, the recombination frequency is 50% since 2 of the 4 gametes were recombinant gametes.
The recombination frequency will be 50% when two genes are located on different chromosomes or when they are widely separated on the same chromosome. This is a consequence of independent assortment.
When two genes are close together on the same chromosome, they do not assort independently and are said to be linked. Whereas genes located on different chromosomes assort independently and have a recombination frequency of 50%, linked genes have a recombination frequency that is less than 50%.
As an example of linkage, consider the classic experiment by William Bateson and Reginald Punnett. They were interested in trait inheritance in the sweet pea and were studying two genes—the gene for flower colour (P, purple, and p, red) and the gene affecting the shape of pollen grains (L, long, and l, round). They crossed the pure lines PPLL and ppll and then self-crossed the resulting PpLl lines. According to Mendelian genetics, the expected phenotypes would occur in a 9:3:3:1 ratio of PL:Pl:pL:pl. To their surprise, they observed an increased frequency of PL and pl and a decreased frequency of Pl and pL (see table below).
Bateson and Punnett experiment
| Phenotype and genotype |
Observed |
Expected from 9:3:3:1 ratio |
| Purple, long (PpLl) |
284 |
216 |
| Purple, round (Ppll) |
21 |
72 |
| Red, long (ppLl) |
21 |
72 |
| Red, round (ppll) |
55 |
24 |
Their experiment revealed linkage between the P and L alleles and the p and l alleles. The frequency of P occurring together with L and with p occurring together with l is greater than that of the recombinant Pl and pL. The recombination frequency cannot be computed directly from this experiment, but intuitively it is less than 50%.
The progeny in this case received two dominant alleles linked on one chromosome (referred to as coupling or cis arrangement). However, after crossover, some progeny could have received one parental chromosome with a dominant allele for one trait (e.g. Purple) linked to a recessive allele for a second trait (e.g. round) with the opposite being true for the other parental chromosome (e.g. red and Long). This is referred to as repulsion or a trans arrangement. The phenotype here would still be purple and long but a test cross of this individual with the recessive parent would produce progeny with much greater proportion of the two crossover phenotypes. While such a problem may not seem likely from this example, unfavorable repulsion linkages do appear when breeding for disease resistance in some crops.
When two genes are located on the same chromosome, the chance of a crossover producing recombination between the genes is related to the distance between the two genes. Thus, the use of recombination frequencies has been used to develop linkage maps or genetic maps.
However, it is important to note that recombination frequency tends to underestimate the distance between two linked genes. This is because as the two genes are located farther apart, the chance of double or even number of crossovers between them also increases. Double or even number of crossovers between the two genes results in them being cosegregated to the same gamete, yielding a parental progeny instead of the expected recombinant progeny.
Meiosis Indicators
With very large pedigrees or with very dense genetic marker data, such as from whole-genome sequencing, it is possible to precisely locate and quantify recombinations. With this type of genetic analysis, a meiosis indicator is assigned to each position of the genome for each meiosis in a pedigree. The indicator indicates which copy of the parental chromosome contributes to the transmitted gamete at that position. For example, if the allele from the 'first' copy of the parental chromosome is transmitted, a '0' might be assigned to that meiosis. If the allele from the 'second' copy of the parental chromosome is transmitted, a '1' would be assigned to that meiosis. The two alleles in the parent came, one each, from two grandparents. These indicators are then used to determine identical-by-descent (IBD) states or inheritance states, which are in turn used to identify genes responsible for diseases and phenotypes.
See also
- Chromosomal crossover
- Genetic association
- Genetic epidemiology
- Genome-wide association study
- Lander–Green algorithm
- Linkage disequilibrium
- Quantitative trait locus
- Association mapping
- Family based QTL mapping
References
- ^ Derivation of mapping function, from Introduction to Genetic Analysis. Griffiths, A. J. F.; Miller, J. H.; Suzuki, D. T.; Lewontin, R. C.; Gelbart, W. M. New York: W. H. Freeman & Co.; 1999
- ^ Graph of mapping function from compared to idealized 1-1 equivalence of recombination frequency percentage (RF%) to map units.
- ^ Advanced genetics from Dr. Ted Helm's plant science course at North Dakota State University.
- ^ Morton NE (1955). "Sequential tests for the detection of linkage". American Journal of Human Genetics 7 (3): 277–318. PMC 1716611. PMID 13258560. //www.ncbi.nlm.nih.gov/pmc/articles/PMC1716611/.
- Griffiths AJF, Miller JH, Suzuki DT, Lewontin RC, Gelbart WM (1993). "Chapter 5". An Introduction to Genetic Analysis (5th ed.). New York: W.H. Freeman and Company. ISBN 0-7167-2285-2.
- Poehlman JM, Sleper DA (1995). "Chapter 3". Breeding Field Crops (4th ed.). Iowa: Iowa State Press. ISBN 0-8138-2427-3.
External links
- Genetic Mapping
- A list of computer programs for genetic analysis including linkage analysis
- Linkage versus Linkage-Disequilibrium
|
Personal genomics
|
|
| Data collection |
- Biobank
- Biological database
|
|
| Field concepts |
- Biological specimen
- De-identification
- Human genetic variation
- Genetic linkage
- Single-nucleotide polymorphisms
- Genetic disorder
|
|
| Applications |
- Personalized medicine
- Predictive medicine
- Genetic epidemiology
|
|
| Analysis techniques |
- Whole genome sequencing
- Genome-wide association study
- SNP array
|
|
| Major projects |
- Human Genome Project
- International HapMap Project
|
|