WordNet
- a pattern of symptoms indicative of some disease
- a complex of concurrent things; "every word has a syndrome of meanings"
PrepTutorEJDIC
- (疾患の徴候となる一群の)症徴候,症候群 / (事件・社会的状態などのパターンを示す)徴候形態
Wikipedia preview
出典(authority):フリー百科事典『ウィキペディア(Wikipedia)』「2023/03/30 11:16:26」(JST)
wiki en
Inflammatory disorder
Medical condition
| Behçet's disease | |
|---|---|
| Other names |
|
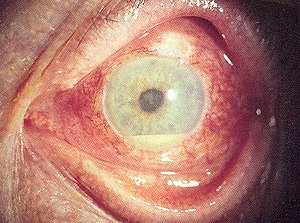 | |
| A person with hypopyon which can be seen in anterior uveitis in a person with Behçet's disease | |
| Pronunciation |
|
| Specialty | Rheumatology, Immunology |
| Symptoms | Mouth sores, genital sores, inflammation of the eye, arthritis,[1] chronic fatigue |
| Complications | Blindness, joint inflammation, blood clots, aneurysm[2] |
| Usual onset | 20s to 40s[1][2] |
| Duration | Long term[1] |
| Causes | Unknown[1] |
| Diagnostic method | Based on symptoms[1] |
| Differential diagnosis | Reactive arthritis, Stevens–Johnson syndrome, Sweet syndrome[2] |
| Medication | Immunosuppressive medication such as corticosteroids[1] |
| Prognosis | Often improves with time[2] |
| Frequency | Rare (US, EU), more common (Middle East, Asia)[2] |
Behçet's disease (BD) is a type of inflammatory disorder which affects multiple parts of the body.[2] The most common symptoms include painful sores on the mucous membranes of the mouth and other parts of the body, inflammation of parts of the eye, and arthritis.[1][2] The sores can last from a few days, up to a week or more.[2] Less commonly there may be inflammation of the brain or spinal cord, blood clots, aneurysms, or blindness.[1][2] Often, the symptoms come and go.[1]
The cause is unknown.[1] It is believed to be partly genetic.[2] Behçet's is not contagious.[1] Diagnosis is based on at least three episodes of mouth sores in a year together with at least two of the following: genital sores, eye inflammation, skin sores, a positive skin prick test.[1]
There is no cure.[1] Treatments may include immunosuppressive medication such as corticosteroids and lifestyle changes.[1] Lidocaine mouthwash may help with the pain.[2] Colchicine may decrease the frequency of attacks.[2]
While rare in the United States and Europe, it is more common in the Middle East and Asia.[2] In Turkey, for example, about 2 per 1,000 are affected.[2] Onset is usually in a person's 20s or 40s.[1][2] The disease was initially described by Turkish dermatologist Hulusi Behçet in 1937.[3]
Signs and symptoms
Skin and mucosa
Nearly all people with Behçet's disease present with some form of painful ulcerations inside the mouth.[4] They are a form of aphthous ulcers or non-scarring oral lesions.[4] The oral lesions are similar to those found in inflammatory bowel disease and can be relapsing.[4] Painful genital ulcerations usually develop around the anus, vulva, or scrotum and cause scarring in 75 percent of the patients.[4] Additionally, patients may present with erythema nodosum, cutaneous pustular vasculitis, and lesions similar to pyoderma gangrenosum.[4]
Eyes
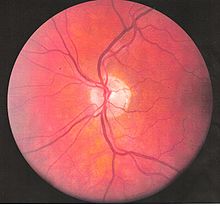
Funduscopic photo left eye centered on the optic disc
Inflammatory eye disease can develop early in the disease course and lead to permanent vision loss in 20 percent of cases. Ocular involvement can be in the form of posterior uveitis, anterior uveitis, or retinal vasculitis. Anterior uveitis presents with painful eyes, conjuctival redness, hypopyon, and decreased visual acuity, while posterior uveitis presents with painless decreased visual acuity and visual field floaters. A rare form of ocular (eye) involvement in this syndrome is retinal vasculitis which presents with painless decrease of vision with the possibility of floaters or visual field defects.[4]
Optic nerve involvement in Behçet's disease is rare, typically presenting as progressive optic atrophy and visual loss. However, cases of acute optic neuropathy (specifically anterior ischemic optic neuropathy) have also been reported to occur.[5] Optic nerve atrophy has been identified as the most common cause of visual impairment. Behçet's disease may result in primary or secondary optic nerve involvement. Papilledema as a result of dural sinus thrombosis[6] and atrophy resulting from retinal disease, have been characterized as secondary causes of optic nerve atrophy in Behçet's disease.[7][8]
Signs and symptoms of acute optic neuropathy include painless loss of vision which may affect either one or both eyes, reduced visual acuity, reduced color vision, relative afferent pupillary defect, central scotoma, swollen optic disc, macular edema, or retrobulbar pain. When these symptoms occur with concurrent mucocutaneous ulcerations, they raise suspicion of acute optic neuropathy in Behçet's Disease. Progressive optic atrophy may result in decreased visual acuity or color vision. Intracranial hypertension with papilledema may be present.[9]
Episcleritis may occur, which causes eye redness and mild pain, without a significant impact on vision.[10]
Bowels
Gastrointestinal (GI) manifestations include abdominal pain, nausea, and diarrhea with or without blood, and they often involve the ileocecal valve.[4] Some patients with BD experience abdominal tenderness, bloating, and general abdominal discomfort. When mild this can resemble irritable bowel syndrome; more severe cases bear similarities to inflammatory bowel diseases such as ulcerative colitis or Crohn's.[citation needed]
Lungs
Lung involvement is typically in the form of hemoptysis, pleuritis, cough, or fever, and in severe cases can be life-threatening if the outlet pulmonary artery develops an aneurysm which ruptures causing severe vascular collapse and death from bleeding in the lungs.[4] Nodules, consolidations, cavities and ground glass lesions are common in patients with pulmonary involvement.[11] Pulmonary artery thrombosis may occur.
Joints
Arthritis is seen in up to half of people, and is usually a non-erosive poly or oligoarthritis primarily of the large joints of the lower extremities.[4]
Brain
Main article: Neuro-Behçet's disease
Central nervous system (CNS) involvement most often occurs as a chronic meningoencephalitis. Lesions tend to occur in the brainstem, the basal ganglia and deep hemispheric white matter and may resemble those of multiple sclerosis (MS). Brainstem atrophy is seen in chronic cases.[citation needed]
Neurological involvements range from aseptic meningitis to vascular thrombosis such as dural sinus thrombosis and organic brain syndrome manifesting with confusion, seizures, and memory loss. Sudden hearing loss (sensorineural) is often associated with it.[4] They often appear late in the progression of the disease but are associated with a poor prognosis.[citation needed]
Heart
Pericarditis is a frequent cardiac manifestation.[11] Chronic aortic regurgitation due to aortic root disease may also be seen.[12] Although infrequent, myocardial infarction (heart attack) with angiographically identified acute coronary artery thrombosis has been reported, including one case with a pathologically demonstrable lesion due to arteritis found at autopsy.[13]
Blood vessels
Blood vessel problems are observed in 7–29% of people with arterial lesions representing 15% of vascular lesions. Arterial lesions pose a greater risk. Most common arterial lesions are occlusions or stenosis and aneurysms or pseudoaneurysms.[14][citation needed]
Cause
The cause is not well-defined, but it is primarily characterized by auto-inflammation of the blood vessels. Although sometimes erroneously referred to as a diagnosis of exclusion, the diagnosis can sometimes be reached by pathologic examination of the affected areas.[15]
The primary mechanism of the damage is autoimmune, which by definition is an overactive immune system that targets the patient's own body. The involvement of a subset of T cells (Th17) seems to be important.[11] The primary cause is not well known. In fact, no one knows yet why the immune system starts to behave this way in Behçet's disease. There does however seem to be a genetic component involved, as first degree relatives of the affected patients are often affected in more than the expected proportion for the general population.[citation needed]
Research suggests that previous infections may provoke the autoimmune responses present in Behçet's disease. Heat shock proteins (HSPs) are present in some bacteria and serve as a "danger signal" to the immune system. However, some HSPs share a similarity in bacteria and humans.[16] The anti-HSP60 and anti-HSP65 antibodies that target HSPs produced by Streptococci (including S. sanguinis and S. pyogenes) and Mycobacterium tuberculosis can also target human HSPs, leading to immune responses linked to uveitis and various symptoms shown in parenchymal neuro-Behçet's disease.[17]
An association with the GIMAP ("GTPase of the immunity-associated protein") family of genes on the long arm of chromosome 7 (7q36.1) has been reported.[18] The genes implicated were GIMAP1, GIMAP2 and GIMAP4.[citation needed]
Pathophysiology

HLA-B51 is strongly associated with Behçet's disease[19]
Behçet's disease is considered more prevalent in the areas surrounding the old silk trading routes in the Middle East and in Central Asia. Thus, it is sometimes known as Silk Road disease. However, this disease is not restricted to people from these regions. A large number of serological studies show a linkage between the disease and HLA-B51.[20] HLA-B51 is more frequently found from the Middle East to South Eastern Siberia, but the incidence of B51 in some studies was 3 fold higher than the normal population. However, B51 tends not to be found in disease when a certain SUMO4 gene variant is involved,[21] and symptoms appear to be milder when HLA-B27 is present.[22] At the current time, a similar infectious origin has not yet been confirmed that leads to Behçet's disease, but certain strains of S. sanguinis has been found to have a homologous antigenicity.[23]
Vasculitis resulting in occlusion of the vessels supplying the optic nerve may be the cause of acute optic neuropathy and progressive optic atrophy in Behçet's disease. Histological evaluation in a reported case of acute optic neuropathy demonstrated substitution of the axonal portion of the optic nerve with fibrous astrocytes without retinal changes.[8] CNS involvement in Behçet's disease may lead to intracranial hypertension most commonly due to dural venous sinus thrombosis[6] and subsequent secondary optic atrophy.
Diagnosis
There is no specific pathological testing or technique available for the diagnosis of the disease, although the International Study Group criteria for the disease are highly sensitive and specific, involving clinical criteria and a pathergy test.[4][24] Behçet's disease has a high degree of resemblance to diseases that cause mucocutaneous lesions such as Herpes simplex labialis, and therefore clinical suspicion should be maintained until all the common causes of oral lesions are ruled out from the differential diagnosis.[citation needed]
Visual acuity, or color vision loss with concurrent mucocutaneous lesions or systemic Behçet's disease symptoms should raise suspicion of optic nerve involvement in Behçet's disease and prompt a work-up for Behçet's disease if not previously diagnosed in addition to an ocular work-up. Diagnosis of Behçet's disease is based on clinical findings including oral and genital ulcers, skin lesions such as erythema nodosum, acne, or folliculitis, ocular inflammatory findings and a pathergy reaction. Inflammatory markers such ESR, and CRP may be elevated. A complete ophthalmic examination may include a slit lamp examination, optical coherence tomography to detect nerve loss, visual field examinations, fundoscopic examination to assess optic disc atrophy and retinal disease, fundoscopic angiography, and visual evoked potentials, which may demonstrate increased latency. Optic nerve enhancement may be identified on Magnetic Resonance Imaging (MRI) in some patients with acute optic neuropathy. However, a normal study does not rule out optic neuropathy. Cerebrospinal fluid (CSF) analysis may demonstrate elevated protein level with or without pleocytosis. Imaging including angiography may be indicated to identify dural venous sinus thrombosis as a cause of intracranial hypertension and optic atrophy.[citation needed]
Diagnostic guidelines

Magnetic resonance venogram demonstrating occlusion of the left sigmoid and transverse sinuses
According to the International Study Group guidelines, for a patient to be diagnosed with Behçet's disease,[24] the patient must have oral (aphthous) ulcers (any shape, size, or number at least 3 times in any 12 months period) along with 2 out of the following 4 "hallmark" symptoms:[citation needed]
- eye inflammation (iritis, uveitis, retinal vasculitis, cells in the vitreous)
- genital ulcers (including anal ulcers and spots in the genital region and swollen testicles or epididymitis in men)
- pathergy reaction (papule >2 mm dia. 24–48 hrs or more after needle-prick). The pathergy test has a specificity of 95 percent to 100 percent, but the results are often negative in American and European patients[4]
- skin lesions (papulo-pustules, folliculitis, erythema nodosum, acne in post-adolescents not on corticosteroids)
Despite the inclusive criteria set forth by the International Study Group, there are cases where not all the criteria can be met and therefore a diagnosis cannot readily be made. There is however a set of clinical findings that a physician can rely upon in making a tentative diagnosis of the disease; essentially Behçet's disease does not always follow the International Study Group guidelines and so a high degree of suspicion for a patient who presents having any number of the following findings is necessary:[citation needed]
- arthritis/arthralgia
- cardio-vascular problems of an inflammatory origin
- changes of personality, psychoses
- deep vein thrombosis
- epididymitis
- extreme exhaustion - chronic fatigue
- inflammatory problems in chest and lungs
- mouth ulcers
- nervous system symptoms
- problems with hearing or balance
- stomach or bowel inflammation
- superficial thrombophlebitis
- any other members of the family with a diagnosis of Behçet's disease.
Treatment
Current treatment is aimed at easing the symptoms, reducing inflammation, and controlling the immune system. The quality of the evidence for treating the oral ulcers associated with Behçet's disease, however, is poor.[25]
High-dose corticosteroid therapy is often used for severe disease manifestations.[26] Anti-TNF therapy such as infliximab has shown promise in treating the uveitis associated with the disease.[27][28] Another Anti-TNF agent, etanercept, may be useful in people with mainly skin and mucosal symptoms.[29] Apremilast may also be used to treat oral ulcers associated with Behçet's disease.[30]
Interferon alpha-2a may also be an effective alternative treatment, particularly for the genital and oral ulcers[31] as well as ocular lesions.[32] Azathioprine, when used in combination with interferon alpha-2b also shows promise,[33] and colchicine can be useful for treating some genital ulcers, erythema nodosum, and arthritis.[34] Benzathine‐penicillin may also reduce new arthritic attacks.[35]
Thalidomide has also been used due to its immune-modifying effect.[36] Dapsone and rebamipide have been shown, in small studies, to have beneficial results for mucocutaneous lesions.[37][38]
Given its rarity, the optimal treatment for acute optic neuropathy in Behçet's disease has not been established. Early identification and treatment are essential. Response to ciclosporin, periocular triamcinolone, and IV methylprednisone followed by oral prednisone has been reported although relapses leading to irreversible visual loss may occur even with treatment.[39] Immunosuppressants such as interferon-alpha and tumour necrosis factor antagonists may improve though not completely reverse symptoms of ocular Behçet's disease, which may progress over time despite treatment. When symptoms are limited to the anterior chamber of the eye prognosis is improved. Posterior involvement, particularly optic nerve involvement, is a poor prognostic indicator. Secondary optic nerve atrophy is frequently irreversible. Lumbar puncture or surgical treatment may be required to prevent optic atrophy in cases of intracranial hypertension refractory to treatment with immunomodulators and steroids.[citation needed]
IVIG could be a treatment for severe[40] or complicated cases.[41][42]
Surgery
Surgical treatment of arterial manifestations of BD bears many pitfalls since the obliterative endarteritis of vasa vasorum causes thickening of the medial layer and splitting of elastin fibers. Therefore, anastomotic pseudoaneurysms are likely to form, as well as pseudoaneurysms at the site of the puncture in case of angiography or endovascular treatment; furthermore, early graft occlusion may occur.[citation needed]
For these reasons, invasive treatment should not be performed in the acute and active phases of the disease when inflammation is at its peak. The evaluation of disease's activity is usually based on relapsing symptoms, ESR (erythrocyte sedimentation rate), and serum levels of CRP (C‐reactive protein).[citation needed]
Endovascular treatment can be an effective and safe alternative to open surgery, with less postoperative complications, faster recovery time, and reduced need for intensive care, while offering patency rates and procedural success rates comparable with those of surgery. This notwithstanding, long‐term results of endovascular treatment in BD are still to be determined.[citation needed]
Epidemiology
The syndrome is rare in the United States, Africa and South America, but is common in Asia, suggesting a possible cause endemic to those areas.[43] A theory suggested that past exposure to lethal infectious agents might have fixed the genetic susceptibility factors to Behçet's disease in those area.[44] An estimated 15,000 to 20,000 Americans have been diagnosed with this disease. In the UK, it is estimated to have about 1 case for every 100,000 people.[45] Globally, males are affected more frequently than females.[46]
In an epidemiologic study, 56 percent of patients with Behçet's disease developed ocular involvement at a mean age of 30.[47] Ocular involvement was the first manifestation of Behçet's disease in 8.6 percent of patients.[47] Ocular Behçet's disease with involvement of the optic nerve is rarely reported. Among patients with ocular Behçet's disease funduscopic findings of optic atrophy, and optic disc paleness have been identified with a frequency of 17.9 percent and 7.4 percent, respectively. Other fundoscopic findings include vascular sheathing (23.7%),[7] retinal hemorrhage (9%),[7] macular edema (11.3%),[7] branch retinal vein occlusion (5.8%),[7] and retinal edema (6.6%).[7] However, optic atrophy was the most significant cause of visual impairment identified in 54 percent of patients with ocular Behçet's disease and permanent visual impairment.[7]
The prevalence of this disease increases from North to South. It follows a more severe course in patients with an early age of onset particularly in patients with eye and gastrointestinal involvement.[48]
Pregnancy
With Behçet's disease as a pre-existing disease in pregnancy or acquired, the pregnancy does not have an adverse effect on the course of Behçet's disease and may possibly ameliorate its course.[49][50] Still, there is a substantial variability in clinical course between patients and even for different pregnancies in the same patient.[49] Also, the other way around, Behçet's disease confers an increased risk of pregnancy complications, miscarriage and Cesarean section.[50]
Behçet's can cause male infertility, either as a result of the condition itself or of a side effect of concomitant medication such as colchicine, which is known to lower sperm count.[51]
History
The first modern formal description of the symptoms was made by H. Planner and F. Remenovsky and published in 1922 in the Archiv für Dermatologie und Syphilis.[52] Behçet's disease is named after Hulusi Behçet (1889–1948), the Turkish dermatologist and scientist who first recognized the three main symptoms of the syndrome in one of his patients in 1924 and reported his research on the disease in Journal of Skin and Venereal Diseases in 1936.[52][53] The name (Morbus Behçet) was formally adopted at the International Congress of Dermatology in Geneva in September 1947. Symptoms of this disease may have been described by Hippocrates in the 5th century BC, in his Epidemion (book 3, case 7).[54]
Some sources use the term "Adamantiades's syndrome" or "Adamantiades–Behçet syndrome", for the work done by Benediktos Adamantiades.[55] However, the current World Health Organization/ICD-10 standard is "Behçet's disease". In 1991, Saudi Arabian medical researchers described neuro-Behçet's disease,[56] a neurological involvement in Behçet's disease, considered one of the most devastating manifestations of the disease.[57] The mechanism can be immune-mediated or thrombotic.[58] The term dates back to at least 1990.[59]
References
- ^ a b c d e f g h i j k l m n o Fleming, Ray (November 2014). "Fast Facts About Behçet's Disease". www.niams.nih.gov. Archived from the original on 13 May 2017. Retrieved 29 May 2017.
- ^ a b c d e f g h i j k l m n o "Behçet's Syndrome". NORD (National Organization for Rare Disorders). 2015. Archived from the original on 11 February 2017. Retrieved 29 May 2017.
- ^ Ball, Gene V.; Fessler, Barri J.; Bridges, S. Louis Jr. (2014). Oxford Textbook of Vasculitis (3 ed.). OUP Oxford. p. 491. ISBN 9780191667022. Archived from the original on 10 September 2017.
- ^ a b c d e f g h i j k l Bolster MB (2009). MKSAP 15 Medical Knowledge Self-assessment Program: Rheumatology. Philadelphia: American College of Physicians. ISBN 978-1-934465-30-1.
- ^ Eye (7 January 2011). "Access : A case of anterior ischemic optic neuropathy associated with Behçet's disease : Eye". Eye. Nature.com. 25 (3): 395–396. doi:10.1038/eye.2010.208. PMC 3178306. PMID 21212799.
- ^ a b Fujikado T, Imagawa K (1994). "Dural sinus thrombosis in Behçet's disease – a case report". Jpn. J. Ophthalmol. 38 (4): 411–16. PMID 7723211.
- ^ a b c d e f g Ozdal PC, Ortaç S, Taşkintuna I, Firat E (2002). "Posterior segment involvement in ocular Behçet's disease". Eur J Ophthalmol. 12 (5): 424–31. doi:10.1177/112067210201200514. PMID 12474927. S2CID 13428859.
- ^ a b Kansu T, Kirkali P, Kansu E, Zileli T (December 1989). "Optic neuropathy in Behçet's disease". J Clin Neuroophthalmol. 9 (4): 277–80. PMID 2531168.
- ^ Behçet syndrome. Yazıcı, Yusuf., Hatemi, Gulen., Seyahi, Emire., Yazici, Hasan. (2nd ed.). Cham: Springer. 2020. p. 77. ISBN 978-3-030-24131-5. OCLC 1127393738.
{{cite book}}: CS1 maint: others (link) - ^ Schonberg, S; Stokkermans, TJ (January 2020). "Episcleritis". PMID 30521217.
{{cite journal}}: Cite journal requires|journal=(help) - ^ a b c Hatemi G, Seyahi E, Fresko I, Hamuryudan V (2012). "Behçet's syndrome: a critical digest of the recent literature". Clin Exp Rheumatol
- ^ Adam C. Ring (23 August 2012). Steven S. Agabegi; Elizabeth Agabegi (eds.). Step-up to medicine (3rd ed.). Philadelphia: Wolters Kluwer/Lippincott Williams & Wilkins. p. 266. ISBN 978-1-60913-360-3.
- ^ Schiff, Steven; Moffatt, Roy; Mandel, William J.; Rubin, Stanley A. (1982). "Acute myocardial infarction and recurrent ventricular arrhythmias in Behcet's syndrome". American Heart Journal. 103 (3): 438–440. doi:10.1016/0002-8703(82)90289-7. PMID 7064781.
- ^ ElGuindy MS, ElGuindy AM (October 2017). "Aneurysmal coronary artery disease: An overview". Global Cardiology Science and Practice. Glob Cardiol Sci Pract. 2017 (3): e201726. doi:10.21542/gcsp.2017.26. PMC 5856968. PMID 29564347.
- ^ "Behcet Disease: Overview – eMedicine Dermatology". Archived from the original on 16 February 2009. Retrieved 28 March 2009.
- ^ Direskeneli, H. (2013). "Innate and Adaptive Responses to Heat Shock Proteins in Behcet's Disease". Genetics Research International. 2013: 249157. doi:10.1155/2013/249157. ISSN 2090-3154. PMC 3893747. PMID 24490075.
- ^ Tanaka, T.; Yamakawa, N.; Koike, N.; Suzuki, J.; Mizuno, F.; Usui, M. (June 1999). "Behçet's disease and antibody titers to various heat-shock protein 60s". Ocular Immunology and Inflammation. 7 (2): 69–74. doi:10.1076/ocii.7.2.69.4018. ISSN 0927-3948. PMID 10420201.
- ^ Lee YJ, Horie Y, Wallace GR, Choi YS, Park JA, Choi JY, Song R, Kang YM, Kang SW, Baek HJ, Kitaichi N, Meguro A, Mizuki N, Namba K, Ishida S, Kim J, Niemczyk E, Lee EY, Song YW, Ohno S, Lee EB (1 September 2013). "Genome-wide association study identifies GIMAP as a novel susceptibility locus for Behcet's disease". Annals of the Rheumatic Diseases. 72 (9): 1510–6. doi:10.1136/annrheumdis-2011-200288. PMID 23041938. S2CID 206848143.
- ^ Ohno S, Ohguchi M, Hirose S, Matsuda H, Wakisaka A, Aizawa M (September 1982). "Close association of HLA-Bw51 with Behçet's disease". Arch. Ophthalmol. 100 (9): 1455–8. doi:10.1001/archopht.1982.01030040433013. PMID 6956266.
- ^ Durrani K, Papaliodis GN (2008). "The genetics of Adamantiades–Behcet's disease". Semin Ophthalmol. 23 (1): 73–9. doi:10.1080/08820530701745264. PMID 18214795. S2CID 11695492.
- ^ Hou S, Yang P, Du L, Zhou H, Lin X, Liu X, Kijlstra A (October 2008). "SUMO4 gene polymorphisms in Chinese Han patients with Behcet's disease". Clin. Immunol. 129 (1): 170–5. doi:10.1016/j.clim.2008.06.006. PMID 18657476.
- ^ Ahn JK, Park YG (October 2007). "Human leukocyte antigen B27 and B51 double-positive Behçet uveitis". Arch. Ophthalmol. 125 (10): 1375–80. doi:10.1001/archopht.125.10.1375. PMID 17923546.
- ^ Yanagihori H, Oyama N, Nakamura K, Mizuki N, Oguma K, Kaneko F (July 2006). "Role of IL-12B promoter polymorphism in Adamantiades-Behcet's disease susceptibility: An involvement of Th1 immunoreactivity against Streptococcus Sanguinis antigen". J. Invest. Dermatol. 126 (7): 1534–40. doi:10.1038/sj.jid.5700203. PMID 16514412.
- ^ a b International Study Group for Behçet's Disease (May 1990). "Criteria for diagnosis of Behçet's disease". Lancet. 335 (8697): 1078–80. doi:10.1016/0140-6736(90)92643-V. PMID 1970380. S2CID 53261546.
- ^ Taylor J, Glenny AM, Walsh T, Brocklehurst P, Riley P, Gorodkin R, Pemberton MN (25 September 2014). Taylor J (ed.). "Interventions for the management of oral ulcers in Behçet's disease". The Cochrane Database of Systematic Reviews. 9 (9): CD011018. doi:10.1002/14651858.CD011018.pub2. PMC 6872426. PMID 25254615.
- ^ CMDT (Current Medical Diagnosis & Treatment) 2007, Chapter 20, page 872
- ^ Sfikakis PP, Theodossiadis PG, Katsiari CG, Kaklamanis P, Markomichelakis NN (2001). "Effect of infliximab on sight-threatening panuveitis in Behcet's disease". Lancet. 358 (9278): 295–6. doi:10.1016/S0140-6736(01)05497-6. PMID 11498218. S2CID 23347314.
- ^ Sfikakis PP (2002). "Behçet's disease: a new target for anti-tumour necrosis factor treatment". Ann Rheum Dis. 61 Suppl 2 (Suppl 2): ii51–3. doi:10.1136/ard.61.suppl_2.ii51. PMC 1766720. PMID 12379622.
- ^ Melikoglu M, Fresko I, Mat C, Ozyazgan Y, Gogus F, Yurdakul S, Hamuryudan V, Yazici H (2005). "Short-term trial of etanercept in Behcet's disease: a double blind, placebo controlled study". J Rheumatol. 32 (1): 98–105. PMID 15630733.
- ^ "Search Orphan Drug Designations and Approvals". www.accessdata.fda.gov. Retrieved 19 November 2019.
- ^ Alpsoy E, Durusoy C, Yilmaz E, Ozgurel Y, Ermis O, Yazar S, Basaran E (2002). "Interferon alfa-2a in the treatment of Behcet disease: a randomized placebo-controlled and double-blind study". Arch Dermatol. 138 (4): 467–71. doi:10.1001/archderm.138.4.467. PMID 11939808.
- ^ Kötter I, Zierhut M, Eckstein AK, Vonthein R, Ness T, Günaydin I, Grimbacher B, Blaschke S, Meyer-Riemann W, Peter HH, Stübiger N (2003). "Human recombinant interferon alpha-2a for the treatment of Behçet's disease with sight-threatening posterior or panuveitis". Br J Ophthalmol. 87 (4): 423–31. doi:10.1136/bjo.87.4.423. PMC 1771623. PMID 12642304.
- ^ Hamuryudan V, Ozyazgan Y, Fresko Y, Mat C, Yurdakul S, Yazici H (2002). "Interferon alpha combined with azathioprine for the uveitis of Behcet's disease: an open study". Isr Med Assoc J. 4 (11 Suppl): 928–30. PMID 12455182.
- ^ Yurdakul S, Mat C, Tüzün Y, Ozyazgan Y, Hamuryudan V, Uysal O, Senocak M, Yazici H (2001). "A double-blind trial of colchicine in Behcet's syndrome". Arthritis Rheum. 44 (11): 2686–92. doi:10.1002/1529-0131(200111)44:11<2686::AID-ART448>3.0.CO;2-H. PMID 11710724. S2CID 19523919.
- ^ Saenz, Antonio; Ausejo, Monica; Shea, Beverley; Wells, George A; Welch, Vivian; Tugwell, Peter (27 April 1998). "Pharmacotherapy for Behcet's syndrome". Cochrane Database of Systematic Reviews. 1998 (2): CD001084. doi:10.1002/14651858.cd001084. ISSN 1465-1858. PMC 7032645. PMID 10796413.
- ^ Hamuryudan V, Mat C, Saip S, Ozyazgan Y, Siva A, Yurdakul S, Zwingenberger K, Yazici H (1998). "Thalidomide in the treatment of the mucocutaneous lesions of the Behcet syndrome. A randomized, double-blind, placebo-controlled trial". Ann Intern Med. 128 (6): 443–50. doi:10.7326/0003-4819-128-6-199803150-00004. PMID 9499327. S2CID 12089634.
- ^ Matsuda T, Ohno S, Hirohata S, Miyanaga Y, Ujihara H, Inaba G, Nakamura S, Tanaka S, Kogure M, Mizushima Y (2003). "Efficacy of rebamipide as adjunctive therapy in the treatment of recurrent oral aphthous ulcers in patients with Behcet's disease: a randomised, double-blind, placebo-controlled study". Drugs in R&D. 4 (1): 19–28. doi:10.2165/00126839-200304010-00002. PMID 12568631. S2CID 23711352.
- ^ Sharquie KE, Najim RA, Abu-Raghif AR (2002). "Dapsone in Behcet's disease: a double-blind, placebo-controlled, cross-over study". The Journal of Dermatology. 29 (5): 267–79. doi:10.1111/j.1346-8138.2002.tb00263.x. PMID 12081158. S2CID 23356719.
- ^ Voros GM, Sandhu SS, Pandit R (2006). "Acute optic neuropathy in patients with Behçet's disease. Report of two cases". Ophthalmologica. 220 (6): 400–05. doi:10.1159/000095869. PMID 17095888. S2CID 45084854.
- ^ Seider N, Beiran I, Scharf J, Miller B (November 2001). "Intravenous immunoglobulin therapy for resistant ocular Behçet's disease". Br J Ophthalmol. 85 (11): 1287–8. doi:10.1136/bjo.85.11.1287. PMC 1723778. PMID 11673289.
- ^ Shutty B, Garg KJ, Swender D, Chernin L, Tcheurekdjian H, Hostoffer R (July 2012). "Optimal use of ivig in a patient with Behçet syndrome and common variable immunodeficiency". Ann. Allergy Asthma Immunol. 109 (1): 84. doi:10.1016/j.anai.2012.05.014. PMID 22727170.
- ^ Beales IL (December 1998). "Gastrointestinal involvement in Behçet's syndrome". Am. J. Gastroenterol. 93 (12): 2633. doi:10.1111/j.1572-0241.1998.02633.x. PMID 9860455. S2CID 26407172.
- ^ "Behcet's Syndrome: MedlinePlus". nih.gov. Archived from the original on 4 July 2016. Retrieved 19 September 2016.
- ^ Piga, M; Mathieu, A (2014). "The origin of Behçet's disease geoepidemiology: possible role of a dual microbial-driven genetic selection". Clinical and Experimental Rheumatology. 32 (4 Suppl 84): S123–9. PMID 24447390.
- ^ Choices, N. H. S. "Behçet's disease – NHS Choices". www.nhs.uk. Archived from the original on 3 March 2016. Retrieved 19 September 2016.
- ^ Escudier M, Bagan J, Scully C (March 2006). "Number VII Behçet's disease (Adamantiades syndrome)". Oral Dis. 12 (2): 78–84. doi:10.1111/j.1601-0825.2005.01144.x. PMID 16476027.
- ^ a b Krause L, Köhler AK, Altenburg A, Papoutsis N, Zouboulis CC, Pleyer U, Stroux A, Foerster MH (May 2009). "Ocular involvement in Adamantiades-Behçet's disease in Berlin, Germany". Graefes Arch. Clin. Exp. Ophthalmol. 247 (5): 661–6. doi:10.1007/s00417-008-0983-4. PMID 18982344. S2CID 23191896.
- ^ Hatemi G, Seyahi E, Fresko I, Hamuryudan V (2013). "Behçet's syndrome: a critical digest of the 2012–2013 literature". Clin Exp Rheumatol. 31 (3 Suppl 77): 108–117. PMID 24064024.
- ^ a b Uzun S, Alpsoy E, Durdu M, Akman A (2003). "The clinical course of Behçet's disease in pregnancy: A retrospective analysis and review of the literature". The Journal of Dermatology. 30 (7): 499–502. doi:10.1111/j.1346-8138.2003.tb00423.x. PMID 12928538. S2CID 12860697.
- ^ a b Jadaon J, Shushan A, Ezra Y, Sela HY, Ozcan C, Rojansky N (2005). "Behcet's disease and pregnancy". Acta Obstetricia et Gynecologica Scandinavica. 84 (10): 939–944. doi:10.1111/j.0001-6349.2005.00761.x. PMID 16167908. S2CID 22363654.
- ^ Behçet's disease – Treatment Archived 5 April 2014 at the Wayback Machine from National Health Service. Page last reviewed: 22 August 2012
- ^ a b synd/1863 at Who Named It?
- ^ Behçet H (1937). "Über rezidivierende, aphtöse, durch ein Virus verursachte Geschwüre am Mund, am Auge und an den Genitalien". Dermatologische Wochenschrift, Hamburg. 105 (36): 1152–1163. Reproduced in Viggor SF, Willis AM, Jawad AS (2011). "Behçet's disease". Grand Rounds. 11: L1–L2. doi:10.1102/1470-5206.2011.L001. Archived from the original on 27 January 2012.
- ^ Verity DH, Wallace GR, Vaughan RW, Stanford MR (29 April 2003). "Behçet's disease: from Hippocrates to the third millennium". Br J Ophthalmol. 87 (9): 1175–83. doi:10.1136/bjo.87.9.1175. PMC 1771837. PMID 12928293.
- ^ B. Adamandiades. Sur un cas d'iritis à hypopyon récidivant. Annales d'oculistique, Paris, 1931, 168: 271–278.
- ^ Malhotra Ravi (2004). "Saudi Arabia". Practical Neurology. 4 (3): 184–185. doi:10.1111/j.1474-7766.2004.03-225.x.
- ^ Saleem S (2005). "Neuro-Behçet's Disease: NBD". Neurographics. 4 (2): 1. Archived from the original on 16 March 2008.
- ^ Al-Araji A, Kidd DP (February 2009). "Neuro-Behçet's disease: epidemiology, clinical characteristics, and management". Lancet Neurol. 8 (2): 192–204. doi:10.1016/S1474-4422(09)70015-8. PMID 19161910. S2CID 16483117.
- ^ Su SL, Way LJ, Lin RT, Peng MJ, Wu SC (March 1990). "Neuro-Behçet's disease: report of three cases with a review of the literature". Gaoxiong Yi Xue Ke Xue Za Zhi. 6 (3): 155–62. PMID 2188002.
Further reading
- Yamauchi Y, Cruz JM, Kaplan HJ, Goto H, Sakai J, Usui M (2005). "Suspected simultaneous bilateral anterior ischemic optic neuropathy in a patient with Behçet's disease". Ocul. Immunol. Inflamm. 13 (4): 317–25. doi:10.1080/09273940590950945. PMID 16159724. S2CID 24830133.
- Brissaud P, Laroche L, de Gramont A, Krulik M (March 1985). "Digital angiography for the diagnosis of dural sinus thrombosis in Behçet's disease". Arthritis Rheum. 28 (3): 359–60. doi:10.1002/art.1780280323. PMID 3884020.
- el-Ramahi KM, al-Kawi MZ (September 1991). "Papilloedema in Behçet's disease: value of MRI in diagnosis of dural sinus thrombosis". J. Neurol. Neurosurg. Psychiatry. 54 (9): 826–29. doi:10.1136/jnnp.54.9.826. PMC 1014525. PMID 1955903.
External links
| Classification | D
|
|---|---|
| External resources |
|
 Medicine portal
Medicine portal
Wikimedia Commons has media related to Behçet's disease.
- Behçet's disease at Curlie
- Questions and answers about Behçet's disease – US National Institute of Arthritis and Musculoskeletal and Skin Diseases
Systemic connective tissue disorders | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| General |
| ||||||||
| Other hypersensitivity/autoimmune |
| ||||||||
| Other |
| ||||||||
Oral and maxillofacial pathology | |||||
|---|---|---|---|---|---|
| |||||
| |||||
| |||||
| |||||
| |||||
| |||||
| |||||
| |||||
| |||||
| |||||
| |||||
|
| Authority control: National |
|
|---|
UpToDate Contents
全文を閲覧するには購読必要です。 To read the full text you will need to subscribe.
- 1. ベーチェット症候群の臨床症状と診断clinical manifestations and diagnosis of behcet syndrome [show details]…oral aphthae of Behcet syndrome; thus, inflammatory bowel disease must be considered before making the diagnosis of Behcet syndrome. The clinical course of intestinal Behcet syndrome is variable. In …
- 2. ベーチェット症候群の治療treatment of behcet syndrome [show details]…rare in Behcet syndrome ), which results from chronic inflammation, require therapy for Behcet syndrome. The preferred therapy is colchicine (1 to 1.2 mg daily), which treats the Behcet syndrome and thus …
- 3. ベーチェット症候群の発症機序pathogenesis of behcet syndrome [show details]…deformability in Behcet syndrome . Tissue-type plasminogen activator levels are lower in patients with deep venous thrombosis who have Behcet syndrome than in those who do not have Behcet syndrome; this suggests …
- 4. 学会が公開する診療ガイドラインのリンク:ベーチェット症候群society guideline links behcet syndrome [show details]…for the management of Behcet syndrome, update (2018) [In English] Japanese National Research Committee for Behcet Disease: Recommendations for the management of neuro-Behcet disease (2020) …
- 5. 血管炎に伴う消化器病変の概要overview of gastrointestinal manifestations of vasculitis [show details]…antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis, polyarteritis nodosa (PAN), and Behcet syndrome, although it can be seen with any of the vasculitides . In three series with a total of 351 patients …
English Journal
- The right place of interleukin-1 inhibitors in the treatment of Behçet's syndrome: a systematic review.
- Bettiol A, Silvestri E, Di Scala G, Amedei A, Becatti M, Fiorillo C, Lopalco G, Salvarani C, Cantarini L, Soriano A, Emmi G.
- Rheumatology international. 2019 Jun;39(6)971-990.
- Behçet's syndrome (BS) is a chronic (auto)-inflammatory disorder characterized by different clusters of symptoms, including mucocutaneous and ocular involvements. Interleukin-1 inhibitors anakinra (ANA), canakinumab (CAN), and gevokizumab (GEV) represent a promising therapeutic alternative in BS. T
- PMID 30799530
- Vikse J, Johnsen SJA, Rønning B, Wildhagen K, Bryne K, Omdal R.
- Tidsskrift for den Norske laegeforening : tidsskrift for praktisk medicin, ny raekke. 2019 May;139(8).
- Uveitis and acute renal failure can be seen in various immune-mediated systemic diseases. Here we present a case of a young man with a rare inflammatory oculorenal syndrome. A man in his thirties was admitted with a constellation of fatigue, flank pain, weight loss and bilateral acute anterior uveit
- PMID 31062556
- Coexistence of Gaucher Disease and severe congenital neutropenia.
- Kose MD, Canda E, Kağnıcı M, Uçar SK, Onay H, Yıldırım Sozmen E, Karapınar D, Özkınay F, Çoker M.
- Blood cells, molecules & diseases. 2019 05;76()1-6.
- Gaucher Disease (GD) is the most common lysosomal storage disorder has traditionally been classified into three clinical phenotypes. Type 3 GD is characterized by neurological involvement but neurological symptoms generally appear later in life than in type 2 disease. Neutropenia is much rarer than
- PMID 30473482
Related Links
- 1.ベーチェット病とはどのような病気ですか. ベーチェット病 (Behçet’s disease)は口腔粘膜のアフタ性潰瘍、外陰部潰瘍、皮膚症状、眼症状の4つの症状を主症状とする慢性再発性の全身性 炎症性疾患 です。. トルコのイスタンブール大学皮膚科Hulsi Behçet教授 ...
- Hatemi G,et al.:Apremilast for Behcet`s syndrome:Abstract from EULAR Annual European Congress of Rheumatology,2018 水木伸久ら,ほか:ベーチェット病診療ガイドライン2020.厚生労働省 難治性疾患政策研究事業 ベーチェット病に関する調査研究班
- Behcet's (beh-CHETS) disease, also called Behcet's syndrome, is a rare disorder that causes blood vessel inflammation throughout your body. The disease can lead to numerous signs and symptoms that can seem unrelated at first.
★リンクテーブル★
| リンク元 | 「ベーチェット病」 |
| 拡張検索 | 「Adamantiades-Behcet syndrome」「neuro-Behcet syndrome」 |
| 関連記事 | 「syndrome」 |
「ベーチェット病」
- 英
- Behçet disease, Behcet disease, Behcet's disease, BD
- 同
- ベーチェット症候群 Behcet syndrome, (国試)Behcet病
- 関
概念
- 難病であり、特定疾患治療研究事業の対象疾患である。
- 急性の炎症が反復し、増悪と寛解を繰り返す慢性疾患であり、繰り返す口内アフタが特徴的である。その他、性器潰瘍、皮膚病変、眼病変、神経病変、血管病変、消化管病変、関節病変を伴う。全身の血管(動静脈)にそのサイズに関係なく炎症を起こすのが特徴的である(参考2)。
病因
- 遺伝要因(HLA-B51)と環境要因が発症に関与。
疫学
- 20歳代に初発。中年男性では多発する。日本人に多い。(NDE.145)
症状
- 特殊型は予後が悪い。
主症状:4主徴
- 1. 口腔内アフタ性潰瘍:ほぼ全例に出現。初発症状で最多。口腔粘膜、舌。有痛性。再発性。
- 2. 陰部潰瘍:70-80%に出現。有痛性の深い潰瘍。男性は陰嚢、女性は大小陰唇。
- 3. 皮膚症状:下腿に好発する結節性紅斑。毛嚢炎様皮疹。血栓性静脈炎(高頻度らしい(NDE.145))など。 皮膚症状の出現頻度は90%(QB.F-147)
- 4. 眼症状:40-50%。男性に多い。網膜ぶどう膜炎、虹彩毛様体炎(前眼房蓄膿)。霧視羞明、眼痛。
副症状
- 5. 関節症状:関節炎症状(腫脹、疼痛)。大関節に多く、骨の破壊や変形は起きない傾向。
- 6. 精巣上体炎:10%以下。腫脹と疼痛。
特殊型
- 7. 消化器症状:回盲部潰瘍。回盲部を中心とした腹痛、下痢、黒色便など。腸Behcet病
- 難治性のことが多い。
- 8. 血管炎症状:大血管の動脈瘤や閉塞。脈拍や血圧の左右差。脳、腎などの循環障害症状。無症状の場合もありうる。血管Behcet病
- 9. 中枢神経症状:髄膜炎症状から、四肢麻痺、痙攣、運動失調、精神症状の出現など。神経Behcet病
ベーチェット病による血管疾患
- 参考1
- 728名のベーチェット病患者における血管疾患
| 疾患名 | 患者数 | |
| 静脈疾患 | 深部静脈血栓症 | 221 |
| 皮下血栓性静脈炎 | 205 | |
| 上大静脈閉塞 | 122 | |
| 下大静脈閉塞 | 93 | |
| 脳静脈洞血栓症 | 30 | |
| バッド・キアリ症候群 | 17 | |
| その他の静脈閉塞 | 24 | |
| 動脈疾患 | 肺動脈閉塞/肺動脈瘤 | 36 |
| 動脈瘤 | 17 | |
| 四肢動脈閉塞/四肢動脈瘤 | 45 | |
| その他の動脈閉塞/動脈瘤 | 42 | |
| 右室血栓症 | 2 | |
検査
- 針反応:皮膚を針で刺すと膿疱ができる。皮膚の被刺激性、好中球機能過剰による
診断
治療
- 眼症状
- 前部ぶどう膜炎:ステロイド点眼、散瞳薬
- 後部ぶどう膜炎:ステロイド結膜下注射/テノン嚢下注射
- 眼炎症予防:コルヒチン、炎症が起きればNSAIDを用いる。
- 難治例ではシクロスポリンを投与、インフリキシマブ投与
症例
参考
- 1. [charged] Clinical manifestations and diagnosis of Behçet’s disease - uptodate [1]
- 2. [charged] Pathogenesis of Behçet’s disease - uptodate [2]
- 3. [charged] Treatment of Behçet’s disease - uptodate [3]
- 4. ベーチェット病 - 難病情報センター
「Adamantiades-Behcet syndrome」
「neuro-Behcet syndrome」
「syndrome」
- n.