| Sideroblastic anemia |
|
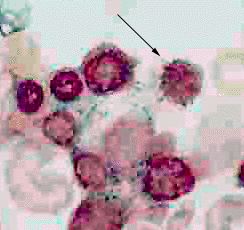
| A ring sideroblast visualized by Prussian blue stain |
| Specialty |
Hematology |
Sideroblastic anemia or sideroachrestic anemia is a form of anemia in which the bone marrow produces ringed sideroblasts rather than healthy red blood cells (erythrocytes).[1] In sideroblastic anemia, the body has iron available but cannot incorporate it into hemoglobin, which red blood cells need in order to transport oxygen efficiently. The disorder may be caused either by a genetic disorder or indirectly as part of myelodysplastic syndrome,[2] which can develop into hematological malignancies (especially acute myeloid leukemia).
Sideroblasts (sidero- + -blast) are atypical, abnormal nucleated erythroblasts (precursors to mature red blood cells) with granules of iron accumulated in the mitochondria surrounding the nucleus.[3] Normally, sideroblasts are present in the bone marrow, and enter the circulation after maturing into a normal erythrocyte.
Ring sideroblasts are named so because iron-laden mitochondria form a ring around the nucleus. To count a cell as a ring sideroblast, the ring must encircle a third or more of the nucleus and contain five or more iron granules, according to the 2008 WHO classification of the tumors of the hematopoietic and lymphoid tissues.[4]
The WHO International Working Group on Morphology of MDS (IWGM-MDS) defined three types of sideroblasts:
- Type 1 sideroblasts: fewer than 5 siderotic granules in the cytoplasm
- Type 2 sideroblasts: 5 or more siderotic granules, but not in a perinuclear distribution
- Type 3 or ring sideroblasts: 5 or more granules in a perinuclear position, surrounding the nucleus or encompassing at least one third of the nuclear circumference.
Contents
- 1 Classification
- 2 Symptoms
- 3 Causes
- 4 Diagnosis
- 5 Treatment
- 6 Course and prognosis
- 7 See also
- 8 References
- 9 External links
Classification
Sideroblastic anemia is typically divided into subtypes based on its cause.
- Hereditary or congenital sideroblastic anemia may be X-linked[5] or autosomal.
| OMIM |
Name |
Gene |
| 300751 |
X-linked sideroblastic anemia (XLSA) |
ALAS2 |
| 301310 |
sideroblastic anemia with spinocerebellar ataxia (ASAT) |
ABCB7 |
| 205950 |
pyridoxine-refractory autosomal recessive sideroblastic anemia |
SLC25A38 |
| 206000 |
pyridoxine-responsive sideroblastic anemia |
(vitamin B6 deficiency; pyridoxal phosphate required for heme synthesis) |
GLRX5 has also been implicated.[6]
- Acquired, or secondary, sideroblastic anemia develops after birth and is divided according to its cause.
Symptoms
Symptoms of sideroblastic anemia include skin paleness, fatigue, dizziness, and enlarged spleen and liver. Heart disease, liver damage, and kidney failure can result from iron buildup in these organs.[7]
Causes
Causes of sideroblastic anemia can be categorized into three groups: congenital sideroblastic anemia, acquired clonal sideroblastic anemia, and acquired reversible sideroblastic anemia. All cases involve dysfunctional heme synthesis or processing. This leads to granular deposition of iron in the mitochondria that form a ring around the nucleus of the developing red blood cell. Congenital forms often present with normocytic or microcytic anemia while acquired forms of sideroblastic anemia are often normocytic or macrocytic.
- Congenital sideroblastic anemia
- X-linked sideroblastic anemia: This is the most common congenital cause of sideroblastic anemia and involves a defect in ALAS2,[8] which is involved in the first step of heme synthesis. Although X-linked, approximately one third of patients are women due to skewed X-inactivation (lyonizations).
- Autosomal recessive sideroblastic anemia involves mutations in the SLC25A38 gene. The function of this protein is not fully understood, but it is involved in mitochondrial transport of glycine. Glycine is a substrate for ALAS2 and necessary for heme synthesis. The autosomal recessive form is typically severe in presentation.
- Genetic syndromes: Rarely, sideroblastic anemia may be part of a congenital syndrome and present with associated findings, such as ataxia, myopathy, and pancreatic insufficiency.
- Acquired clonal sideroblastic anemia
- Clonal sideroblastic anemias fall under the broader category of myelodysplastic syndromes (MDS). Three forms exist and include refractory anemia with ringed sideroblasts (RARS), refractory anemia with ringed sideroblasts and thrombocytosis (RARS-T), and refractory cytopenia with multilineage dysplasia and ringed sideroblasts (RCMD-RS). These anemias are associated with increased risk for leukemic evolution.
- Acquired reversible sideroblastic anemia
- Causes include excessive alcohol use (the most common cause of sideroblastic anemia), pyridoxine deficiency, lead poisoning, and copper deficiency. Excess zinc[9] can indirectly cause sideroblastic anemia by decreasing absorption and increasing excretion of copper. Antimicrobials that may lead to sideroblastic anemia include isoniazid, chloramphenicol, cycloserine, and linezolid.[10]
Diagnosis
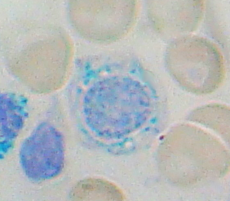
Bone marrow aspirate: ring sideroblasts
Ringed sideroblasts are seen in the bone marrow.
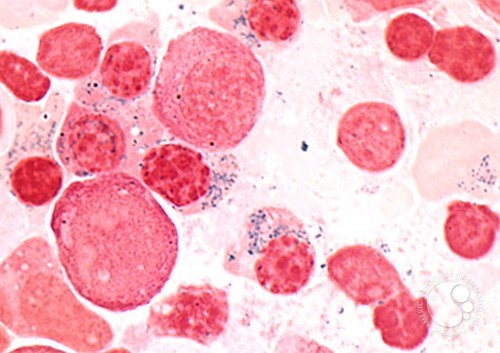
The anemia is moderate to severe and dimorphic. Microscopic viewing of the red blood cells will reveal marked unequal cell size and abnormal cell shape. Basophilic stippling is marked and target cells are common. Pappenheimer bodies are present in the red blood cells. The mean cell volume is commonly decreased (i.e., a microcytic anemia), but MCV may also be normal or even high. The RDW is increased with the red blood cell histogram shifted to the left. Leukocytes and platelets are normal. Bone marrow shows erythroid hyperplasia with a maturation arrest.
In excess of 40% of the developing erythrocytes are ringed sideroblasts. Serum iron, percentage saturation and ferritin are increased. The total iron-binding capacity of the cells is normal to decreased. Stainable marrow hemosiderin is increased.
Laboratory findings
- Serum Iron: High
- Increased ferritin levels
- Normal total iron-binding capacity
- High transferrin saturation
- Hematocrit of about 20-30%
- The mean corpuscular volume or MCV is usually normal or low for congenital causes of sideroblastic anemia but normal or high for acquired forms.
- With lead poisoning, see coarse basophilic stippling of red blood cells on peripheral blood smear
- Specific test: Prussian blue stain of RBC in marrow shows ringed sideroblasts. Prussian blue staining involves a non-enzymatic reaction of ferrous iron with ferrocyanide forming ferric-ferrocyanide, which is blue in color. A counterstain may be used to provide better visualization.
Treatment
Occasionally, the anemia is so severe that support with transfusion is required. These patients usually do not respond to erythropoietin therapy.[11] Some cases have been reported that the anemia is reversed or heme level is improved through use of moderate to high doses of pyrodoxine (vitamin B6). In severe cases of SBA, bone marrow transplant is also an option with limited information about the success rate. Some cases are listed on MedLine and various other medical sites. In the case of isoniazid-induced sideroblastic anemia, the addition of B6 is sufficient to correct the anemia. Desferrioxamine, a chelating agent, is used to treat iron overload from transfusions. Therapeutic phlebotomy can be used to manage iron overload.[12]
Course and prognosis
Sideroblastic anemias are often described as responsive or non-responsive in terms of increased hemoglobin levels to pharmacological doses of vitamin B6.
1- Congenital: 80% are responsive, though the anemia does not completely resolve.
2- Acquired clonal: 40% are responsive, but the response may be minimal.
3- Acquired reversible: 60% are responsive, but course depends on treatment of the underlying cause.
Severe refractory sideroblastic anemias requiring regular transfusions and/or that undergo leukemic transformation (5-10%) significantly reduce life expectancy.
See also
- Anemia
- Siderosis
- List of hematologic conditions
- Hematopoietic stem cell transplantation
References
- ^ Caudill JS, Imran H, Porcher JC, Steensma DP (October 2008). "Congenital sideroblastic anemia associated with germline polymorphisms reducing expression of FECH". Haematologica. 93 (10): 1582–4. doi:10.3324/haematol.12597. PMID 18698088.
- ^ Sideroblastic Anemias: Anemias Caused by Deficient Erythropoiesis at Merck Manual of Diagnosis and Therapy Professional Edition
- ^ "Sideroblast" at Dorland's Medical Dictionary
- ^ Mufti, GJ; Bennett, JM; Goasguen, J; Bain, BJ; Baumann, I; Brunning, R; Cazzola, M; Fenaux, P; Germing, U; Hellström-Lindberg, E; Jinnai, I; Manabe, A; Matsuda, A; Niemeyer, CM; Sanz, G; Tomonaga, M; Vallespi, T; Yoshimi, A; International Working Group on Morphology of Myelodysplastic, Syndrome (Nov 2008). "Diagnosis and classification of myelodysplastic syndrome: International Working Group on Morphology of myelodysplastic syndrome (IWGM-MDS) consensus proposals for the definition and enumeration of myeloblasts and ring sideroblasts". Haematologica. 93 (11): 1712–7. doi:10.3324/haematol.13405. PMID 18838480.
- ^ X-linked sideroblastic anemia at NLM Genetics Home Reference
- ^ Camaschella C (September 2008). "Recent advances in the understanding of inherited sideroblastic anaemia". Br. J. Haematol. 143 (1): 27–38. doi:10.1111/j.1365-2141.2008.07290.x. PMID 18637800.
- ^ Genetics Home Reference: Genetic Conditions > X-linked sideroblastic anemia Reviewed October 2006. Retrieved on 5 Mars, 2009
- ^ Aivado M, Gattermann N, Rong A, et al. (2006). "X-linked sideroblastic anemia associated with a novel ALAS2 mutation and unfortunate skewed X-chromosome inactivation patterns". Blood Cells Mol. Dis. 37 (1): 40–5. doi:10.1016/j.bcmd.2006.04.003. PMID 16735131.
- ^ Forman, W.B. (1990). "Zinc abuse: an unsuspected cause of sideroblastic anemia". West J Med. 152: 190–2. PMC 1002314 . PMID 2400417.
- ^ Saini, N; Jacobson, JO; Jha, S; Saini, V; Weinger, R (April 2012). "The perils of not digging deep enough--uncovering a rare cause of acquired anemia". American journal of hematology. 87 (4): 413–6. doi:10.1002/ajh.22235. PMID 22120958.
- ^ Papadakis, Maxine A.; Tierney, Lawrence M.; McPhee, Stephen J. (2005). "Sideroblastic Anemia". Current Medical Diagnosis & Treatment, 2006. McGraw-Hill Medical. ISBN 0-07-145410-1.
- ^ Peto, T. E. A., Pippard, M. J., Weatherall, D. J. Iron overload in mild sideroblastic anaemias" Lancet 321: 375-378, 1983. Note: Originally Volume I.
External links
| Classification |
V · T · D
- ICD-10: D64.0-D64.3
- ICD-9-CM: 285.0
- OMIM: 301310 206000 300751
- MeSH: D000756
- DiseasesDB: 12110
|
- GeneReviews/NCBI/NIH/UW entry on X-Linked Sideroblastic Anemia and Ataxia
|
Diseases of red blood cells (D50–69,74, 280–287)
|
| ↑ |
|
| ↓ |
| Anemia |
| Nutritional |
- Micro-: Iron-deficiency anemia
- Macro-: Megaloblastic anemia
|
Hemolytic
(mostly normo-) |
| Hereditary |
- enzymopathy: G6PD
- glycolysis
- hemoglobinopathy: Thalassemia
- Sickle-cell disease/trait
- HPFH
- membrane: Hereditary spherocytosis
- Minkowski–Chauffard syndrome
- Hereditary elliptocytosis
- Southeast Asian ovalocytosis
- Hereditary stomatocytosis
|
| Acquired |
- Drug-induced autoimmune
- Drug-induced nonautoimmune
- Hemolytic disease of the newborn
|
|
Aplastic
(mostly normo-) |
- Hereditary: Fanconi anemia
- Diamond–Blackfan anemia
- Acquired: PRCA
- Sideroblastic anemia
- Myelophthisic
|
| Blood tests |
- MCV
- Normocytic
- Microcytic
- Macrocytic
- MCHC
|
|
| Other |
- Methemoglobinemia
- Sulfhemoglobinemia
- Reticulocytopenia
|
|
|
Myeloid hematological malignancy/leukemia histology (ICD-O 9590–9989, C81–C96, 200–208)
|
CFU-GM/
and other granulocytes |
| CFU-GM |
| Myelocyte |
| AML: |
- Acute myeloblastic leukemia
- M0
- M1
- M2
- APL/M3
|
| MP |
- Chronic neutrophilic leukemia
|
|
| Monocyte |
| AML |
- AMoL/M5
- Myeloid dendritic cell leukemia
|
| CML |
- Philadelphia chromosome
- Accelerated phase chronic myelogenous leukemia
|
|
| Myelomonocyte |
| AML |
|
| MD-MP |
- Juvenile myelomonocytic leukemia
- Chronic myelomonocytic leukemia
|
|
| Other |
|
|
| CFU-Baso |
|
| CFU-Eos |
| AML |
|
| MP |
- Chronic eosinophilic leukemia/Hypereosinophilic syndrome
|
|
|
| MEP |
| CFU-Meg |
|
| CFU-E |
| AML |
|
| MP |
|
| MD |
- Refractory anemia
- Refractory anemia with excess of blasts
- Chromosome 5q deletion syndrome
- Sideroblastic anemia
- Paroxysmal nocturnal hemoglobinuria
- Refractory cytopenia with multilineage dysplasia
|
|
|
| CFU-Mast |
| Mastocytoma |
- Mast cell leukemia
- Mast cell sarcoma
- Systemic mastocytosis
|
| Mastocytosis: |
- Diffuse cutaneous mastocytosis
- Erythrodermic mastocytosis
- Adult type of generalized eruption of cutaneous mastocytosis
- Urticaria pigmentosa
- Mast cell sarcoma
- Solitary mastocytoma
|
| Systemic mastocytosis |
- Xanthelasmoidal mastocytosis
|
|
| Multiple/unknown |
| AML |
- Acute panmyelosis with myelofibrosis
- Myeloid sarcoma
|
| MP |
- Myelofibrosis
- Acute biphenotypic leukaemia
|
|
|
Sex linkage: X-linked disorders
|
|
X-linked recessive
|
| Immune |
- Chronic granulomatous disease (CYBB)
- Wiskott–Aldrich syndrome
- X-linked severe combined immunodeficiency
- X-linked agammaglobulinemia
- Hyper-IgM syndrome type 1
- IPEX
- X-linked lymphoproliferative disease
- Properdin deficiency
|
| Hematologic |
- Haemophilia A
- Haemophilia B
- X-linked sideroblastic anemia
|
| Endocrine |
- Androgen insensitivity syndrome/Spinal and bulbar muscular atrophy
- KAL1 Kallmann syndrome
- X-linked adrenal hypoplasia congenita
|
| Metabolic |
- Amino acid: Ornithine transcarbamylase deficiency
- Oculocerebrorenal syndrome
- Dyslipidemia: Adrenoleukodystrophy
- Carbohydrate metabolism: Glucose-6-phosphate dehydrogenase deficiency
- Pyruvate dehydrogenase deficiency
- Danon disease/glycogen storage disease Type IIb
- Lipid storage disorder: Fabry's disease
- Mucopolysaccharidosis: Hunter syndrome
- Purine–pyrimidine metabolism: Lesch–Nyhan syndrome
- Mineral: Menkes disease/Occipital horn syndrome
|
| Nervous system |
- X-linked mental retardation: Coffin–Lowry syndrome
- MASA syndrome
- Alpha-thalassemia mental retardation syndrome
- Siderius X-linked mental retardation syndrome
- Eye disorders: Color blindness (red and green, but not blue)
- Ocular albinism (1)
- Norrie disease
- Choroideremia
- Other: Charcot–Marie–Tooth disease (CMTX2-3)
- Pelizaeus–Merzbacher disease
- SMAX2
|
| Skin and related tissue |
- Dyskeratosis congenita
- Hypohidrotic ectodermal dysplasia (EDA)
- X-linked ichthyosis
- X-linked endothelial corneal dystrophy
|
| Neuromuscular |
- Becker's muscular dystrophy/Duchenne
- Centronuclear myopathy (MTM1)
- Conradi–Hünermann syndrome
- Emery–Dreifuss muscular dystrophy 1
|
| Urologic |
- Alport syndrome
- Dent's disease
- X-linked nephrogenic diabetes insipidus
|
| Bone/tooth |
- AMELX Amelogenesis imperfecta
|
| No primary system |
- Barth syndrome
- McLeod syndrome
- Smith–Fineman–Myers syndrome
- Simpson–Golabi–Behmel syndrome
- Mohr–Tranebjærg syndrome
- Nasodigitoacoustic syndrome
|
|
|
X-linked dominant
|
- X-linked hypophosphatemia
- Focal dermal hypoplasia
- Fragile X syndrome
- Aicardi syndrome
- Incontinentia pigmenti
- Rett syndrome
- CHILD syndrome
- Lujan–Fryns syndrome
- Orofaciodigital syndrome 1
- Craniofrontonasal dysplasia
|
|
|
Genetic disorder, membrane: ABC-transporter disorders
|
| ABCA |
- ABCA1 (Tangier disease)
- ABCA3 (Surfactant metabolism dysfunction 3)
- ABCA4 (Stargardt disease 1, Retinitis pigmentosa 19)
- ABCA12 (Harlequin-type ichthyosis, Lamellar ichthyosis 2)
|
| ABCB |
- ABCB4 (Progressive familial intrahepatic cholestasis 3)
- ABCB7 (ASAT)
- ABCB11 (Progressive familial intrahepatic cholestasis 2)
|
| ABCC |
- ABCC2 (Dubin–Johnson syndrome)
- ABCC6 (Pseudoxanthoma elasticum)
- ABCC7 (Cystic fibrosis)
- ABCC8 (HHF1, TNDM2)
- ABCC9 (Dilated cardiomyopathy 1O)
|
| ABCD |
- ABCD1 (Adrenoleukodystrophy, Adrenomyeloneuropathy)
|
| ABCG |
- ABCG5 (Sitosterolemia)
- ABCG8 (Gallbladder disease 4, Sitosterolemia)
|
|
see also ABC transporters
|