| Nail–patella syndrome |
|
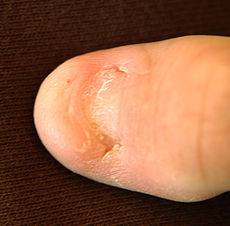
Nail of a patient with nail–patella syndrome
|
| Classification and external resources |
| Specialty |
medical genetics |
| ICD-10 |
Q87.2 (ILDS Q87.230) |
| ICD-9-CM |
756.89 |
| OMIM |
161200 |
| DiseasesDB |
8773 |
| eMedicine |
ped/1546 derm/813 |
| MeSH |
C05.550.629 |
| Orphanet |
2614 |
|
[edit on Wikidata]
|
Nail–patella syndrome (NPS) (also known as "HOOD syndrome") is a genetic disorder that results in small, poorly developed nails and kneecaps, but can also affect many other areas of the body, such as the elbows, chest, and hips. The name "nail–patella" can be very misleading because the syndrome often affects many other areas of the body, including even the production of certain proteins.[1]:666 Those affected by NPS may have one or more affected areas of the body, and its severity varies depending on the individual. It is also referred to as iliac horn syndrome, hereditary onychoosteodysplasia (HOOD syndrome), Fong disease or Turner–Kieser syndrome.[2]
Diagnosis of NPS can be made at birth, but is common for it to remain undiagnosed for several generations. While there is no cure available for NPS, treatment is available and recommended.
Contents
- 1 Genetics
- 2 Symptoms
- 3 Presentation
- 4 Treatment
- 5 See also
- 6 References
- 7 External links
Genetics
Nail–patella syndrome is inherited in an autosomal dominant pattern.
The Nail–patella syndrome is inherited via autosomal dominancy linked to aberrancy on human chromosome 9's q arm (the longer arm), 9q34. This autosomal dominancy means that only a single copy, instead of both, is sufficient for the disorder to be expressed in the offspring, meaning the chance of getting the disorder from an affected heterozygous parent is 50%. The frequency of the occurrence is 1/50,000.[citation needed] The disorder is linked to the ABO blood group locus.
It is associated with random mutations in the LMX1B gene. Studies have been conducted and 83 mutations of this gene have been identified.[3][4][5]
Symptoms
The skeletal structures of individuals who have this disorder may have pronounced deformities. As reported by several medical doctors, the following features are commonly found in people who suffer from nail–patella syndrome:[6]
Bones and joints
AP radiograph showing a hypoplastic patella in NPS
AP radiograph of the right iliac crest showing a bony exostosis or posterior iliac horn, which is pathognomonic of NPS
- Patellar involvement is present in approximately 90% of patients; however, patellar aplasia occurs in only 20%.
- In instances in which the patellae are smaller or luxated, the knees may be unstable.
- The elbows may have limited motion (e.g., limited pronation, supination, extension).
- Subluxation of the radial head may occur.
- Arthrodysplasia of the elbows is reported in approximately 90% of patients.
- General hyperextension of the joints can be present.
- Exostoses arising from the posterior aspect of the iliac bones ("iliac horns") are present in as many as 80% of patients; this finding is considered pathognomonic for the syndrome.
- Other reported bone changes include scoliosis, scapular hypoplasia, and the presence of cervical ribs.
-
An elbow of a man who suffers from nail–patella syndrome (NPS)
-
This is a view from a different angle of the same man's other elbow
Kidney issues may arise such as proteinuria and nephritis. Proteinuria is usually the first sign of renal involvement and either rapidly or years after suffering from asymptomatic proteinuria, renal failure occurs in around 5% of NPS patients. Hypothyroidism, irritable bowel syndrome, attention deficit hyperactivity disorder (ADHD), and thin tooth enamel are associated with NPS, but whether these are related or simply coincidences are unclear.[7]
Presentation
The hallmark features of this syndrome are poorly developed fingernails, toenails, and patellae (kneecaps). Sometimes, this disease causes the affected person to have either no thumbnails or a small piece of a thumbnail on the edge of the thumb. The lack of development, or complete absence of fingernails results from the loss of function mutations in the LMX1B gene. This mutation may cause a reduction in dorsalising signals, which then results in the failure to normally develop dorsal specific structures such as nails and patellae.[8] Other common abnormalities include elbow deformities, abnormally shaped pelvic (hip) bones, and kidney (renal) disease.
Treatment
Treatment for NPS varies depending on the symptoms observed.
- Perform screening for renal disease and glaucoma, surgery, intensive physiotherapy, or genetic counseling.[3]
- ACE inhibitors are taken to treat proteinuria and hypertension in NPS patients.
- Dialysis and renal transplant.
- Physical therapy, bracing and analgesics for joint pain.
- Other surgery treatments such as patella realignment, joint replacement, and the cutting away of the head of radius.
See also
- List of cutaneous conditions
- List of radiographic findings associated with cutaneous conditions
References
- ^ Freedberg, et al. (2003). Fitzpatrick's Dermatology in General Medicine. (6th ed.). McGraw-Hill. ISBN 0-07-138076-0.
- ^ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. Page 786-7. ISBN 0-7216-2921-0.
- ^ a b Sweeney E, Fryer A. "Nail patella syndrome: a review of the phenotype aided by developmental biology". Journal of Medical Genetics. 40 (3): 153–162. doi:10.1136/jmg.2003.
- ^ Towers AL, Clay CA, Sereika SM, McIntosh I, Greenspan SL (April 2005). "Skeletal integrity in patients with nail patella syndrome". J. Clin. Endocrinol. Metab. 90 (4): 1961–5. doi:10.1210/jc.2004-0997. PMID 15623820.
- ^ Romero P., Sanhueza F., Lopez P., Reyes L., Herrera L. (2011). "c.194 A>C (Q65P) mutation in the LMX1B gene in patients with nail-patella syndrome associated with glaucoma". Molecular vision. 17: 1929–39. PMID 21850167.
- ^ Choczaj-Kukula, A., & Janniger, C. K. (2009). Nail–patella syndrome. In emedicine: WebMD. Retrieved October 11, 2009, from WebMD database.
- ^ Buatti Chris (August 2007). "Nail-Patella Syndrome". Consultant 360. 47 (8).
- ^ Wright M J. "Achondroplasia and nail-patella syndrome: the compound phenotype". Journal of Medical Genetics. 37 (9): 25. doi:10.1136/jmg.37.9.e25.2000.
External links
- [1]
- GeneReview/NCBI/NIH/UW entry on nail–patella syndrome
- Rare Disorders: Stargardt's Disease – very detailed article with images
- Nail Patella Syndrome UK
- Nail Patella Syndrome Holland
- Nail–Patella Syndrome – article at National Center for Biotechnology Information
|
Congenital abnormality syndromes (Q87, 759.7)
|
|
| Craniofacial |
- Acrocephalosyndactylia
- Apert syndrome
- Carpenter syndrome
- Pfeiffer syndrome
- Saethre–Chotzen syndrome
- Sakati–Nyhan–Tisdale syndrome
|
|
| other: |
- Baller–Gerold syndrome
- Cyclopia
- Goldenhar syndrome
- Möbius syndrome
|
|
|
| Short stature |
- 1q21.1 deletion syndrome
- Aarskog–Scott syndrome
- Cockayne syndrome
- Cornelia de Lange Syndrome
- Dubowitz syndrome
- Noonan syndrome
- Robinow syndrome
- Silver–Russell syndrome
- Seckel syndrome
- Smith–Lemli–Opitz syndrome
- Turner syndrome
|
|
| Limbs |
- Adducted thumb syndrome
- Holt–Oram syndrome
- Klippel–Trénaunay–Weber syndrome
- Nail–patella syndrome
- Rubinstein–Taybi syndrome
|
|
| Gastrulation/mesoderm: |
- Caudal regression syndrome
- Ectromelia
- Sirenomelia
- VACTERL association
|
|
|
| Overgrowth |
- Beckwith–Wiedemann syndrome
- Perlman syndrome
- Sotos syndrome
- Weaver syndrome
|
|
| Laurence–Moon–Bardet–Biedl |
- Bardet–Biedl syndrome
- Laurence–Moon syndrome
|
|
Combined/other,
known locus |
- 2 (Feingold syndrome)
- 3 (Zimmermann–Laband syndrome)
- 4/13 (Fraser syndrome)
- 8 (Branchio-oto-renal syndrome, CHARGE syndrome)
- 12 (Keutel syndrome, Timothy syndrome)
- 15 (Marfan syndrome)
- 19 (Donohue syndrome)
- Multiple
|
|
Genetic disorder, protein biosynthesis: Transcription factor/coregulator deficiencies
|
|
| (1) Basic domains |
| 1.2 |
- Feingold syndrome
- Saethre–Chotzen syndrome
|
|
| 1.3 |
|
|
|
(2) Zinc finger
DNA-binding domains |
| 2.1 |
- (Intracellular receptor): Thyroid hormone resistance
- Androgen insensitivity syndrome
- Kennedy's disease
- PHA1AD pseudohypoaldosteronism
- Estrogen insensitivity syndrome
- X-linked adrenal hypoplasia congenita
- MODY 1
- Familial partial lipodystrophy 3
- SF1 XY gonadal dysgenesis
|
|
| 2.2 |
- Barakat syndrome
- Tricho–rhino–phalangeal syndrome
|
|
| 2.3 |
- Greig cephalopolysyndactyly syndrome/Pallister–Hall syndrome
- Denys–Drash syndrome
- Duane-radial ray syndrome
- MODY 7
- MRX 89
- Townes–Brocks syndrome
- Acrocallosal syndrome
- Myotonic dystrophy 2
|
|
| 2.5 |
- Autoimmune polyendocrine syndrome type 1
|
|
|
| (3) Helix-turn-helix domains |
| 3.1 |
- ARX
- Ohtahara syndrome
- Lissencephaly X2
- MNX1
- HOXD13
- PDX1
- LMX1B
- MSX1
- Tooth and nail syndrome
- OFC5
- PITX2
- POU4F3
- POU3F4
- ZEB1
- Posterior polymorphous corneal dystrophy
- Fuchs' dystrophy 3
- ZEB2
|
|
| 3.2 |
- PAX2
- PAX3
- PAX4
- PAX6
- Gillespie syndrome
- Coloboma of optic nerve
- PAX8
- Congenital hypothyroidism 2
- PAX9
|
|
| 3.3 |
- FOXC1
- Axenfeld syndrome 3
- Iridogoniodysgenesis, dominant type
- FOXC2
- Lymphedema–distichiasis syndrome
- FOXE1
- Bamforth–Lazarus syndrome
- FOXE3
- Anterior segment mesenchymal dysgenesis
- FOXF1
- FOXI1
- Enlarged vestibular aqueduct
- FOXL2
- Premature ovarian failure 3
- FOXP3
|
|
| 3.5 |
- IRF6
- Van der Woude syndrome
- Popliteal pterygium syndrome
|
|
|
(4) β-Scaffold factors
with minor groove contacts |
| 4.2 |
- Hyperimmunoglobulin E syndrome
|
|
| 4.3 |
- Holt–Oram syndrome
- Li–Fraumeni syndrome
- Ulnar–mammary syndrome
|
|
| 4.7 |
- Campomelic dysplasia
- MODY 3
- MODY 5
- SF1
- SRY XY gonadal dysgenesis
- Premature ovarian failure 7
- SOX10
- Waardenburg syndrome 4c
- Yemenite deaf-blind hypopigmentation syndrome
|
|
| 4.11 |
|
|
|
| (0) Other transcription factors |
|
|
| Ungrouped |
- TCF4
- ZFP57
- TP63
- Rapp–Hodgkin syndrome/Hay–Wells syndrome/Ectrodactyly–ectodermal dysplasia–cleft syndrome 3/Limb–mammary syndrome/OFC8
|
|
| Transcription coregulators |
| Coactivator: |
- CREBBP
- Rubinstein–Taybi syndrome
|
|
| Corepressor: |
- HR (Atrichia with papular lesions)
|
|