- 同
- 神経腫瘍字
Wikipedia preview
出典(authority):フリー百科事典『ウィキペディア(Wikipedia)』「2023/02/09 00:56:49」(JST)
wiki en
Field of study
For the journal, see Neuro-Oncology (journal).
| Focus | Cancerous brain tumors |
|---|---|
| Significant tests | Tumor markers, TNM staging, CT scans, MRI |
| Specialist | Neurooncologist |
Neuro-oncology is the study of brain and spinal cord neoplasms, many of which are (at least eventually) very dangerous and life-threatening (astrocytoma, glioma, glioblastoma multiforme, ependymoma, pontine glioma, and brain stem tumors are among the many examples of these). Among the malignant brain cancers, gliomas of the brainstem and pons, glioblastoma multiforme, and high-grade (highly anaplastic) astrocytoma/oligodendroglioma are among the worst.[1] In these cases, untreated survival usually amounts to only a few months, and survival with current radiation and chemotherapy treatments may extend that time from around a year to a year and a half, possibly two or more, depending on the patient's condition, immune function, treatments used, and the specific type of malignant brain neoplasm. Surgery may in some cases be curative, but, as a general rule, malignant brain cancers tend to regenerate and emerge from remission easily, especially highly malignant cases. In such cases, the goal is to excise as much of the mass (tumor cells) and as much of the tumor margin as possible without endangering vital functions or other important cognitive abilities. The Journal of Neuro-Oncology is the longest continuously published journal in the field and serves as a leading reference to those practicing in the area of neuro-oncology.
General information
Primary tumors of the central nervous system
Primary brain tumors can occur at any age, from infancy to late in life. These tumors often afflict people during their prime years. Factors such as age, tumor location, and clinical presentation are helpful in differential diagnosis. Most types of primary brain tumors are more common in men with the exception of meningiomas, which are more common in women.[2]
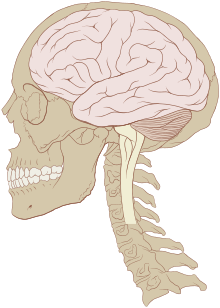
Human Central Nervous System
Metastatic tumors of the central nervous system
Cancer spreads to the nervous system by direct invasion, compression, or metastasis. Direct invasion or compression from continuous tissues relates to the proximity of the nervous system to other structures, such as the brachial plexus, lumbosacral plexus, vertebral neuroforamina, base of skull, cranium, and pelvic bones.[2]
Intracranial metastasis
There are three types of intracranial metastasis: brain metastasis, dural metastasis, and leptomeningeal metastasis. Brain metastasis can be single or multiple and involve any portion of the brain. Metastasis to dural structures generally occurs by hematogenous spread or direct invasion from a contiguous bone. Dural metastases can invade the underlying brain and cause focal edema and associated neurologic symptoms. These processes tend to cause seizures early in the course because of their cortical location. Metastasis to the leptomeninges is an uncommon but well-recognized clinical presentation in cancer patients. Leptomeningeal metastasis most commonly is due to breast, lung, or melanoma primary tumors.[2]
Skull metastasis
Metastases to the skull are divided into two categories by general site: calvarium and skull base. Metastases to the calvarium usually are asymptomatic. Metastases to the skull base quickly become symptomatic because of their proximity to cranial nerves and vascular structures.[2]
Spinal metastasis
The spine most often is affected by metastatic disease involving the epidural space. This usually occurs as direct tumor spread from a vertebral body (85%) or by invasion of paravertebral masses through a neuroforamin (10–15%).[2]
Genetic syndromes and risk factors
There are multiple hereditary conditions that increase a person's chance of developing brain tumors.
Nongenetic risk factors
Few issues in medicine are as potentially contentious as the suspicion of environmental and occupational causes of cancer, including brain tumors. Prior cranial irradiation is the only risk factor that definitely predisposes to brain tumor formation. Some of the risk factors are ionizing radiation, nonionizing radiation, nitrosamines and industrial chemicals.
Mechanisms
Tumor factors
Histology
Seizures are common in patients with low-grade tumors such as dysembryoblastic neuroepithelial tumors, gangligliomas, and oligodendrogliomas. The rapid growth of fast-growing high-grade brain tumors may damage the subcortical network essential for electrical transmission, whereas slow-growing tumors have been suggested to induce partial deafferentation of cortical regions, causing denervation hypersensitivity and producing an epileptogenic milieu. Studies strongly suggest that genetic factors may play a role in tumor development and tumor-related epilepsy.[3][4]
Tumor location
The location of tumors is closely related to their histology. The majority of glioneuronal tumors occur in the temporal lobe. Some data have shown that oligodendroglial tumors were more likely to be located in frontal lobe, whereas astrocytomas were more commonly found in temporal locations. It may be postulated that tumor-related seizures have unique characteristics, which may share some common genetic pathways with tumorigenesis.
Blood-brain barrier disruption (BBB)
Human and animal studies have suggested that perturbations in neurovascular integrity and breakdown of the BBB lead to neuronal hypersynchronization and epileptiform activity. Relevant molecular changes in brain tumors that affect BBB structure and function include decreased expression of transmembrane junctional proteins and heightened release of vascular endothelial growth factor. Results suggest that pathological disruption of the BBB in brain tumor patients may contribute to seizure activity.
Peri-tumoral factors
Contemporary imaging techniques provide testimony to the remarkable differences between the peri-tumoral brain and normal tissue.
Morphological changes
Certain morphological changes in the peri-tumoral brain tissue, such as persistent neurons in the white matter, inefficient neuronal migration, and changes in synaptic vesicles, are also believed to contribute to seizure generation.
Hypoxia, acidosis and metabolic changes
Tumors with insufficient blood supply often cause interstitial hypoxia, which subsequently contributes to acidosis. The intratumoral hypoxia and acidosis may extend to the surrounding tissue. Furthermore, hypoxia causes acidosis as a consequence of both heightened metabolic requirements of the proliferating tissue and impaired oxidative energy metabolism.
Ionic changes
Ionic changes in the peri-tumoral zone may influence neuronal activity. An interesting hypothesis was proposed by Sontheimer, who suggested that glioma invasion into the peri-tumoral zone is in part mediated by chloride channel overexpression, allowing cells to traverse the extracellular space through rapid changes in cell shape.
Glutamate neurotransmission
Recent work has demonstrated a close link between seizure activity and high extracellular glutamate in tumor-related epilepsy. Glutamate activation of ionotropic receptors leads to a rapid excitatory signal based on cation influx that can cause release of calcium from intracellular stores.[2]
Initial patient evaluation and care
1. Brain Tumor Presentations
In general, patients with primary brain tumors or single metastatic tumors can present with any of these signs and symptoms, whereas patients with multiple brain metastases tend to present with generalized symptoms and may lack localized findings.[5]
Several clinical features warrant special comment:
- Seizures (partial or generalized) are the presenting symptom in 15-20% of patients with intracranial tumors. Seizures occur in up to 50% of patients with melanoma metastases, oligodendrogliomas, and tumors that have a hemorrhagic component. Seizures also are more common with cortically based tumors.[5]
- Seizures are much less common in patients with infratentorial tumors than in those with supratentorial tumors.[5]
- "Stroke-like" onset of symptoms is due to hemorrhage within the tumor or, less commonly, macroscopic tumor embolus from systemic cancer.[5]
- Although intratumoral hemorrhage can occur in any primary or metastatic brain tumor, certain tumors have a greater tendency to bleed, including metastasis from melanoma, choriocarcinoma, and thyroid cancer and the primary brain tumors glioblastoma and oligodendroglioma.[5]
2. Spinal Cord Tumor Presentations
- Pain is the first symptom in >90% of patients presenting with epidural metastasis and occurs less frequently with intradural tumors.[6]
- Mechanisms of pain include spinal cord ischemia and traction on the periosteum, dura, nearby soft tissues, and nerve roots.[6]
- Pain occasionally can be absent in adults and more often is absent in childhood. If other neurologic symptoms suggestive of myelopathy are present, without pain, the clinician should evaluate for spinal cord tumor.[6]
- Changes in bowel and bladder habits, particularly urinary retention with overflow incontinence, usually occur late in the course of epidural spinal cord compression but are seen in a small percentage of patients at presentation.[6]
3. Approach to the Evaluation of New Patients
The initial evaluation of a patient with a newly diagnosed tumor of the nervous system is a critical step toward appropriate management and patient care. The most important portions of the initial evaluation are a detailed history and a thorough examination. This process serves to identify the extent and nature of neurological deficit, provides diagnostic clues, can help disclose a source of metastasis, or may identify a genetic process associated with a primary central nervous system tumor.[5]
4. Practical Strategies for Providing Appropriate Patient Care
There is no question that the clinical management of neurooncology patients is challenging. However, if we are to help patients and ultimately make advances in treating these tumors, meticulous and compassionate care of patients with neurological malignancies are crucial.[5]
- Give instructions both orally and in written form for the patient to take home.[5]
- Use a consistent format of written instructions, so that a patient can expect where to find information on the page.[5]
- Write down new or important diagnoses for the patient to refer to at home.[5]
- Identify one reliable caregiver to serve as a contact point.[5]
- Pictures and diagrams are helpful.[5]
- A team approach, using clinicians with different areas of expertise, is helpful.[5]
- Provide a reliable and simple method for the patient to seek help.[5]
- Minimize sedating drug use.[5]
Diagnostic procedures
Diagnostic imaging of the brain and spinal cord
The imaging studies commonly used in neurooncology are computed tomography (CT) and magnetic resonance imaging (MRI). Less commonly used are myelography, positron emission tomography (PET), and diagnostic angiography.[7][8]
Lumbar puncture and cerebrospinal fluid analysis
Lumbar puncture (LP) and cerebrospinal fluid (CSF) analysis are important for the evaluation of some primary tumors, metastatic conditions, and neurologic complications of cancer.[7]
Pathologic diagnosis
Accurate histologic diagnosis is critical for treatment planning and patient counseling. Surgically obtained tissue usually is required to make a histologic diagnosis. For certain tumors, a definitive diagnosis can be accomplished by vitreous aspirate, cerebrospinal fluid (CSF) cytology, or suggested by the presence of certain tumor markers in the CSF.[7]
Commonly used treatments
- Radiotherapy
Radiotherapy is an important treatment for central nervous system tumors and has been demonstrated to extend survival and improve the quality of life for patients with many of the primary and metastatic brain tumors.[7] - Chemotherapy
Chemotherapy, or the use of drugs in the treatment of cancer, can lead to the long-term control of many malignancies. Some tumors, such as testicular cancer of Hodgkin's disease, may be cured even when they are widespread. As chemotherapy may be associated with severe toxicity, it should be given under the supervision of one skilled in the administration and monitoring of such agents.[7] - Corticosteroids
Corticosteroids (CS) are commonly used in patients with a variety of neuro-oncologic conditions. CS treatment often is required to control symptoms related to increased intracranial pressure (ICP) or peritumoral edema.[9] - Neurosurgical interventions
Neurosurgical intervention is warranted in almost all cases of primary central nervous system tumors and for many metastatic tumors. A biopsy usually establishes a definitive histologic diagnosis. The role of surgery depends on the nature of the tumor. With modern neurosurgical techniques, most patients with extra-axial brain tumors are cured with minimal residual neurologic deficit.[9]
Specific tumors
Tumor types
Gliomas
Main article: Glioma
Primary central nervous system (CNS) tumors involve a variety of pathologic tissues, each with its own natural history. Due to the fact that gliomas alone account for almost 40 percent of all CNS tumors, it is common in the literature to distinguish between glial and nonglial tumors.
Astrocytomas
Main article: Astrocytoma
Astrocytoma Various category systems have been proposed in the literature over time for grading the malignancy of astrocytomas. Since 1993, the four-level rating system proposed by the World Health Organization (WHO) has been the most widely used and applied. It is based on four histological features: increased cell density, mitosis, endothelial proliferation and necrosis. Thereafter, grade I astrocytomas, such as pilocytic astrocytomas, are typically benign histology. Astrocytomas of II grade (diffuse) show increased cell density as the only histological feature and are neoplasms with a lower degree of infiltration. Astrocytomas III show a significant mitosis. grade (anaplastic). And endothelial proliferation or necrosis can be seen in grade IV astrocytomas, known as glioblastomas.
Low-grade astrocytomas

Low-grade astrocytoma: A pilocytic astrocytoma of the hypothalamic region
Pilocytic astrocytomas (including pilomyxoid aastrocytoma), subependymal giant cell astrocytomas, and pleomorphic xanthastrocytomas are among the tumors that have been described. These are somewhat rarer neoplasms of benign histology that can often only be cured by surgery. If the excision is incomplete, the remaining tumorous tissue could be successfully treated with radiation therapy. In rare cases where local treatment does not work, systemic chemotherapy can be successful, which must be individually adjusted. Children respond to a combination of carboplatin and vincristine.

MRI of a diffuse astrocytoma of varying degrees. The dark area shows a low grade astrocytoma of the frontal lobe and the two small lighter inner ones higher grade neoplasms.

Microscopic control of the acidic glial fibrillary proteins of the subependymal giant cell astrocytoma. Their monoclonal antibody is used to differentiate primary gliomas from metastatic lesions in the brain and from astrocytes in tumors outside the CNS.
On computed tomography, diffuse grade II astrocytomas appear as less intense lesions. In the preferred magnetic resonance imaging, contrast agents may not be able to highlight these neoplasms, their luminescence may be thinner and weaker. A more intense one may indicate tissues of increased anaplasia. Whenever possible, a biopsy is suggested to obtain samples from the anaplastic portion of the tumor.
In most cases, patients with diffuse astrocytomas are 20 to 40 years old. The occurrence of epileptic seizures is typical for them. Conditions for a favorable prognosis are young age, tumor size below 50 millimeters and the most extensive surgical resection of the tumor possible. Late relapses are relatively common, requiring patients to be followed up for 15 years after tumor removal.
Despite their relatively sluggish course, most astrocytomas progress to lesions characterized by extensive anaplasia that are usually refractory to surgery and radiation therapy. However, the therapy for patients with diffuse low-grade astrocytomas does not show a unanimous consensus in the literature. The role of complete resection is discussed in professional contexts. The results of some studies show that maximum tumor removal gives the best results. In fact, small and unilateral tumors can be completely removed if no critical structures of the brain are involved. A pragmatic approach that is generally acceptable for the generality of cases is to remove the neoplasia as far as possible to avoid significant neurological deficits.
Studies have shown that radiation therapy given immediately after diagnosis has prolonged the time the patient is disease-free before tumor recurrence compared to the situation where the course of radiation therapy is delayed until the time of progression. However, there is currently no consensus that radiation therapy shortly after diagnosis improves the patient's 'overall survival'.
In patients with milder or no symptoms, or with seizures that can be controlled with antiseizure drugs, it is possible to delay radiotherapy until tumor growth reaches a critical phase. There is often a desire to reduce the risk of neurological damage caused by radiation therapy itself.
Two prospective randomized clinical trials showed no greater benefit from high-dose radiotherapy than from low-dose. Typically, the total dosage is between 45 and 54 Gray with a fractionation of 1.8 to 2 Gray.
The effect of adjuvant chemotherapy in patients with low-grade astrocytomas is still under investigation. Preliminary results from a clinical trial comparing radiotherapy alone to radiotherapy followed by chemotherapy containing procarbazine, lomustine, and vincristine (PCV) showed a longer period of "disease-free survival" with the combination, but no prolonged "overall survival." Due to the toxicity associated with the PCV protocol, the use of temozolomide is recommended both as initial therapy and post-recovery.
Anaplastic astrocytomas
Main article: Anaplastic astrocytoma

Magnetic resonance imaging of different therapy phases of an anaplastic astrocytoma
Anaplastic astrocytoma is a malignant brain tumor characterized by diffuse growth, increased cell density, and nuclear division figures. It arises from a specific cell population of the central nervous system, the Astrocytes. According to the WHO classification of tumours of the central nervous system, the tumor corresponds to a grade III tumor. Typically, patients with anaplastic astrocytoma have epileptic seizures, focal neurologicalschematic deficits, headache, and personality changes. The average patient age is 45 years. Magnetic resonance imaging generally shows a massive lesion with an increased contrast[disambiguation needed] signal, which can also be weaker. Diagnosis is by histological examination of the lesion by biopsy or surgical resection. A poorer prognosis may be associated with advanced age, poor physical condition, and significant neurological damage.
In general, the therapeutic outcome is better with complete surgical resection (standard treatment) without increasing neurological deficits. Radiation therapy is standard because it has been shown to increase survival time. The role of chemotherapy is contrversial.
Glioblastoma
Main article: Glioblastoma

Untreated glioblastoma
The most common and most malignant glial cell tumors are glioblastomae. They consist of a heterogeneous mass of poorly differentiated astrocytoma cells mainly in adults. Usually they occur in the hemispheres, more rarely in the brainstem or spinal cord. Except in very rare cases, like all brain tumorss, they do not extend beyond the structures of the central nervous systems.
The glioblastoma can develop from a diffuse (II degree) or a anaplastic astrocytoma (III degree). In the latter case, it is referred to as Sequela. However, when it occurs with no antecedents or evidence of an earlier malignancy, it is referred to as primary. Glioblastomas are treated with surgery, radiation, and chemotherapy. They are difficult to cure and there are few cases surviving beyond three years.
Oligodendrogliomas
Main article: Oligodendroglioma
Oligodendroglioma is an unusual brain tumor of glial cells that arises from oligodendrocytes. It occurs mainly in adults between 40 and 45 years of age, preferentially in the cerebral cortex and the white matter of the cerebral hemispheres. Oligodendrogliomas are relatively rare, accounting for less than about 5 percent of all primary brain tumors and no more than about 10 to 15 percent of all gliomas.
These tumors are divided into low-grade and anaplastic lesions. Anaplastic oligodendroglioma is characterized by increased cell density, mitosis, endothelial proliferation and nuclear polymorphism, and necrosis.
Low-grade oligodendrogliomas and oligoastrocytomas
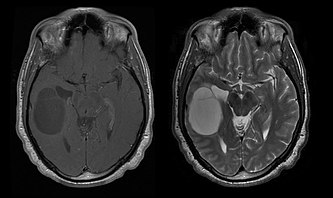
Comparison of horizontal  (left) and
(left) and  magnetic resonance imaging of an oligodendroglioma
magnetic resonance imaging of an oligodendroglioma
(left) and magnetic resonance imaging of an oligodendrogliomaThe median survival for patients with pure oligodendroglioma is about ten years, with oligoastrocytoma about eight years. The lengthening compared to pure astrocytomass is due to a deletion or translocation of the 1p/19q pair in the tumor.
The average age of patients at diagnosis is 35 years. Typical symptoms are epileptic seizures, but focal neurological deficits, personality changes or other symptoms of cranial pressure such as headache and vomiting can also be reported will. These tumors are not usually visible in computed tomography, therefore magnetic resonance imaging is the method of choice for diagnostic imaging. On the
These tumors develop more slowly than low-grade astrocytomas and there is no consensus in the literature regarding optimal treatment. Initial treatment includes symptom control with anticonvulsants, radiation, chemotherapy, or a combination of the latter two. Surgery, radiotherapy and chemotherapy play an important role in relapses. Resections can relieve symptoms. On temozolomide, 50 percent of patients who relapsed after radiation therapy had a positive reaction.
Anaplastic oligodendrogliomas and oligoastrocytomas
Main article: Anaplastic oligodendroglioma
Mathematical modeling of the growth of an anaplastic Oligodendroglioma based on brain topological structures, study of two pones (video)
Anaplastic oligodendroglioma have typical symptoms resulting from mass effect and epileptic seizures. Despite their chemosensitivity, median survival is only 3 to 5 years. Treatment involves maximum excision[disambiguation needed] followed by radiotherapy. With regard to chemotherapy, in two recent phase III clinical trials, the results of radiotherapy were compared with those of combined radiotherapy and procarbazine, lomustine, vincristine chemotherapy. Although the Survival analysis without relevant symptoms was longer with the combined therapy, the overall survival was the same with both therapies. Patients with 1p/19q-deletion achieved the best therapy results, patients without 1p/19q deletion were able to improve their results with PCV chemotherapy.
Prospectivee clinical studyn have shown that approximately 50 to 70 percent of patients with recurrent anaplastic oligodendroglioma after radiotherapy respond positively to chemotherapy with PCV or temozolomide. Although superior efficacy of temozolomide and PCV therapy has not been established, the lack of accumulation Myelosuppression with temozolomide for use at the start of relapse treatment.
Ependymomas
Main article: Ependymom
 - Magnetic resonance axial projection of an ependymomas
- Magnetic resonance axial projection of an ependymomasEpendymoma is a neoplasm that develops from ependymal cells lining the cerebral ventricles, choroid plexus, filum terminale, and central canal of the spinal cord. Ependymal cells are also present in the brain parenchyma as a result of embryonic migration from periventricular areas to the cerebral cortex.
These fairly rare tumors can appear at any age, but they have two characteristic peaks, from 0 to 10 and from 40 to 50 years. Intracranial injuries, which usually occur in the posterior fossa, are more common in the first age group, while spinal injuries are more common in the second age group.
Ependymomas are divided into low-grade lesions (I and II grades on the WHO scale) and anaplastic lesions (III grades). I. grade are in particular subependymomas and myxopapillary ependymomas, III. Anaplastic ependymoma. Patients with low-grade ependymomas in the spine that can be completely removed do not undergo radiation therapy afterwards. The role of postoperative radiotherapy in low-grade intracranial ependymomas is controversial, but radiotherapy treatment is usually indicated for anaplastic or low-grade tumors that cannot be completely resected.
Clinical studies have shown that ependymomas respond to chemotherapy, particularly platinum-based ones. The benefit for platinum-based chemotherapy is 67 percent, compared to 25 percent for nitrosourea. The prognosis for grade II ependymomas is 68 percent 6-year disease-free survival and 87 percent overall survival. In anaplastic ependymomas, these figures drop to 29 percent and 37 percent, respectively.
Nonglial tumors
Medulloblastomas
Main article: Medulloblastoma
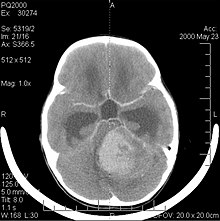
Computed tomography of a medulloblastoma
Medulloblastoma is the most common malignant brain tumor in children. The highest incidence occurs in children between the ages of 2 and 7 years.
The greatest risk of disease remains in childhood, as medulloblastoma is very rare in people over the age of 21. This tumor is typical of the posterior fossa, where it is localized in both hemispheres of the cerebellum or in the cerebellar vermis. Because it is invasive and fast growing, it usually spreads to other parts of the central nervous system (CNS) via the CSF and can infiltrate the floor of the nearby fourth ventricle and the meninges. Rarely, additional CNS metastases can occur. When the malignancy occurs, symptoms include loss of balance, incoordination, diplopia, dysarthria, and due to involvement of the fourth ventricle, which often results in obstructive hydrocephalus, headache, nausea and vomiting, and unstable gait.
MRI usually shows a massive contrast-enhancing lesion involving the cerebellum. As mentioned above, medulloblastoma has a high propensity to locally infiltrate the leptomeninges as well as to spread through the subarachnoid space, involving the ventricles, cerebral convexity, and leptomeningeal surfaces of the spine. Consequently, it is necessary to bring the entire craniospinal axis into resonance.
The purpose of surgery is to remove as much of the mass presented by the lesion as possible. In fact, postoperative residual tumors result in a poorer prognosis. Also a harbinger of an unfavorable prognosis is the presence of tumor cells in the cerebrospinal fluid or the resonance detection of leptomeningeal metastases. Surgery alone is usually not curative. In some cases, however, therapeutic irradiation of the craniospinal axis, focused on the primary tumor site, may result. Adding chemotherapy after radiation therapy increases the cure rate. Platinum-based drugs (cisplatin or carboplatin), etoposide, and an alkylating agent (cyclophosphamide or lomustine) are used with vincristine. With appropriate treatment, long-term survival of more than 3 years in medulloblastoma patients ranges from 60 to 80 percent.
Meningiomas
Main article: Meningioma

Macroscopy of a meningioma: It is clearly visible that the tumor is pressing on the brain instead of infiltrating it
Histology of a meningioma
Meningiomas are the most common intracranial extrinsic or extra-axial brain tumors that arise from the cells of the arachnoid, the membrane that lines the brain and spinal cord. The incidence of this neoplasia is about 2 cases per year per 100,000 population. They are more common in women in their sixth and seventh decades. Their frequency is higher in patients with type 2 neurofibromatosis. Loss of chromosome 22 is characteristic of meningiomas, although the prognostic significance of this finding is unclear.
Patients with meningioma may present with symptoms typical of a massive skull lesion, including seizures and focal neurological deficits. Because meningioma can also be asymptomatic, they are sometimes detected on computed tomography and magnetic resonance imaging for other reasons. This resonance tumor has a characteristic appearance, usually consisting of uniform contrast enhancement along the dura with clear separation from the brain parenchyma.
Another feature, although not present in all cases, is the so-called "dural tail," represented by a bulge that extends beyond the lesion and indicates the anchorage point in the dura Many incidentally discovered meningiomas do not require treatment at the time of initial diagnosis. If the patient is found to have a significant mass effect, whether or not symptoms are present, the treatment of choice is usually complete resection. In a Mayo Clinics studio comparing tumor control rates after surgical resection and radiosurgery in patients with small-to-moderate intracranial meningioma and no symptoms of mass effect, radiosurgery resulted in better control (98 versus 88 percent) and with fewer complications (10 versus 22 percent) compared to surgical removal.
Primary CNS lymphomas

Horizontal magnetic resonance imaging of a primary CNS lymphoma
magnetic resonance imaging of a primary CNS lymphomaPrimary central nervous system lymphomas account for about 2 percent to 3 percent of all brain tumors in patients with a normal immune system. They are more common in men aged 55 to 60. Almost half of all lymphomas occur in patients over the age of 60 and about a quarter in patients over the age of 70. The incidence seems to increase with age, but the reason is still unclear. Patients with a weakened immune system are at higher risk of developing CNS lymphoma, so those who have had an organ transplant have a congenital immunodeficiency or autoimmune disease or are infected with human immunodeficiency virus. HIV-associated brain lymphomas are associated with Epstein-Barr virus, particularly in patients with CD4 lymphocyte counts below 500 cells per cubic millimeter in the blood. Most CNS lymphomas are diffuse large B-cell lymphomas in type.
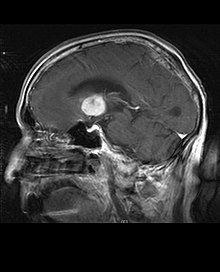 MRI of a primary CNS lymphoma in the sagittal plane
MRI of a primary CNS lymphoma in the sagittal plane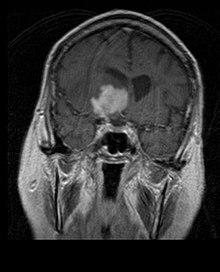
Frontal magnetic resonance imaging of a primary CNS lymphoma
magnetic resonance imaging of a primary CNS lymphomaPatients suffer from a variety of characteristic symptoms of a focal or multifocal massive lesion. MRI usually shows tumors with homogeneous contrast enhancement within the deep periventricular white matter. Multifocality and inhomogeneous enhancement are typical for patients with a weakened immune system. Analysis of CNS lymphoma is extremely important in the differential diagnosis of brain neoplasia.
The administration of corticosteroids can lead to the complete disappearance of the enhancement, which complicates the diagnosis of the lesions. Consequently, if CNS lymphoma is to be considered in the differential diagnosis, corticosteroids should be avoided unless the mass effect causes a serious and immediate problem in the patient.
Biopsy of the suspected lesion is crucial. In contrast to systemic large B-cell lymphoma, in which both chemotherapy and radiotherapy are effective, and treatment of localized lesions is curative, central nervous system lymphoma typically responds to initial therapy but then recurs. As with systemic lymphoma, the role of surgery is primarily limited to obtaining appropriate tissue samples for diagnosis.
In the past, radiation therapy was given to the whole brain (panencephalic). The median survival is about 12 months, even with localized lesions. Recurrence usually affects the site of the previous injury as well as other regions. Responses to chemotherapy are more promising. Clinical trials in which high-dose methotrexate alone was used as the first treatment and radiotherapy was delayed until the time of relapse or progression showed better overall survival than radiotherapy alone. Even more effective was the combination of methotrexate, vincristine, procarbazine, intrathecal methotrexate, cytarabine, and panencephalic radiotherapy and cytarabine, or the use of intra-arterial chemotherapy with intra-arterial methotrexate, intravenously injected cyclophosphamide, and etoposide after modification of the blood-brain barrier with mannitol. The median survival in methotrexate regimens was 24 to 40 months, much higher than that in radiotherapy alone (range 24 to 40 months). In some cases, radiation therapy is only used for relapses when there is an initial regression with chemotherapy.
Cases of long survival have also been reported without radiotherapy. Panencephalic radiation therapy is associated with a high risk of developing dementia or leukoencephalopathy. This risk could be reduced by developing effective tumor control strategies that avoid panencephalic radiotherapy. Initial therapy for patients with compromised immune systems is to reduce the causes of immunosuppression. The prognosis for these patients is usually worse than that for patients who have a normal immune system. Due to accompanying tumor infections and a generally suboptimal physical condition, chemotherapy can often not be carried out in these immunosuppressed patients. As with other brain tumors, response to treatments depends on age and physical condition.
Metastatic tumors of the central nervous system
Brain metastases
Main article: Brain metastasis
Brain metastases are the most common intracranial neoplasms in adults, being ten times more common than primary brain tumors. They occur in 20 to 40 percent of adults with cancer and are mainly associated with lung and breast cancer and melanoma. These lesions result from the spread of cancer cells through the bloodstream and most commonly occur at the junction of gray and white matter, where the cross-section of blood vessels changes, trapping tumor cell embolisms. 80 percent of the lesions occur in the cerebral hemispheres, 15 percent in the cerebellum, and 5 percent in the brainstem. Approximately 80 percent of patients have a history of systemic cancer and 70 percent have multiple brain metastases.

Horizontal MRI of a brain metastasis from a melanoma
MRI of a brain metastasis from a melanomaSignificant advances have recently been made in the diagnosis and management of these lesions, resulting in improved survival and symptom control. The onset of signs and symptoms are similar to those of other massive lesions in the brain. The diagnostic method of choice is magnetic resonance imaging using contrast media.
The literature shows equivalent results for surgery and radiosurgery. The latter appears to be more convenient, effective, and safer for small lesions or in regions inaccessible to surgery. Radiosurgery is a sensible alternative for patients who cannot be operated on for medical reasons. However, surgery is clearly the optimal method to obtain tissues for diagnosis and to remove the lesions that cause mass effect. Therefore, radiosurgery and surgery should better be considered as two complementary but different methods to be applied depending on the different situation of the patient. For nearly 50 percent of patients with one or two brain metastases, surgical removal is not an option because of inaccessibility of the lesions, extent of systemic disease, or other factors. These and other patients with multiple metastases are usually offered panencephalic radiation therapy as a standard of care. In fact, up to almost 50 percent of them achieve an improvement in neurological symptoms and 50 to 70 percent a noticeable response with this therapy. Chemotherapy is rarely used primarily for brain metastases.
For most patients with brain metastases, median survival is only four to six months after panencephalic radiation therapy.
However, patients younger than 60 years with discrete lesions and controlled systemic disease may achieve longer survival because they can tolerate a more aggressive treatment approach.
Meningeal metastases

Histogram of meningeal metastases
Metastases of the soft meninges (leptomeninges encephali) can be diagnosed in about 5 percent of tumor patients. Most often they occur in melanoma, breast and lung cancer as a result of the spread of tumor cells through the bloodstream. The malignant cells are then spread throughout the central nervous system (CNS), generally via the liquor cerebrospinalis, colloquially also known as brain fluid.
One or more of the following signs and symptoms may be caused by meningeal metastases, among others:
- local nerve damage such as cranial nerve paralysis, motor weakness and radiculopathies, paresthesia and pain,
- direct invasion of the brain or spinal tissue,
- Disorder of the blood vessels in the brain and spine with focal neurological deficits and/or seizures,
- Impediments to the normal flow of cerebrospinal fluid with headache and increased intracranial pressure,
- Disorders of normal brain function such as encephalopathy and/or perivascular infiltration by tumor cells with resulting ischemia and apoplexy symptoms.
Diagnosis can be made by examination of the cerebrospinal fluid or magnetic resonance imaging of the brain and spinal cord. The presence of malignant cells can be detected in 50 percent of patients. Cytological examination remains negative in at least 10 percent of patients with leptomeningeal involvement. Increasing the number of lumbar punctures up to six and the volume of fluid removed to 10 milliliters per puncture can increase the possibility of a positive diagnosis. In the cerebrospinal fluid, the concentration of proteins is usually high, that of glucose can be low in the presence of pleocytosis. Radiographic study may show hydrocephalus without a massive lesion or diffuse enlargement of the leptomeninges.
Without therapy, median survival is 4 to 6 weeks, with death attributed to progressive neurological deterioration. Leptomeningeal metastases are often a manifestation of the end-stage of the main disease, and symptomatic therapy may be the most appropriate solution. Corticosteroids and analgesics provide temporary relief. Treatment may be offered to patients with minimal systemic disease and acceptable general physical condition to relieve symptoms and prolong survival.
Median survival can be increased from 3 to 6 months with radiotherapy to symptomatic sites and more voluminous diseased areas identified by x-ray, and intrathecal therapy with methotrexate, cytarabine, and thiotepa performed with lumbar puncture or Ommaya catheter.
The major complication of methotrexate-based intrathecal therapy is necrotizing leukoencephalopathy, which can develop after months of therapy in those few patients who can enjoy prolonged survival. This devastating toxic effect is particularly common in patients who have received prior or concomitant radiotherapy with intrathecal methotrexate therapy.
Clinical problems encountered in neuro-oncology
This section does not cite any sources. Please help improve this section by adding citations to reliable sources. Unsourced material may be challenged and removed. (January 2021) (Learn how and when to remove this template message) |
- anorexia and weight loss
- brain tumors in women of childbearing age
- central nervous system infections
- constipation
- cranial nerve syndromes
- deep venous thrombosis and pulmonary embolus
- depression and anxiety
- differential diagnosis of brain tumor progression
- fatigue and weakness
- fever and neutropenia
- gait disturbances
- headaches
- hiccups
- increased intracranial pressure, brain herniation syndromes, and coma
- insomnia
- mental status changes
- nausea and vomiting
- paraneoplastic syndromes
- peripheral nerve problems: plexopathy and neuropathy
- seizures and other spells
- stroke and other cerebrovascular complications
- urinary problems
- visual symptoms
Pain and terminal care
Palliative care is a special type of care provided to improve the quality of life of patients who have a serious or life-threatening disease, such as cancer. The purpose of palliative care is not to cure but to prevent or treat, as early as possible, the symptoms and side effects of the disease and its treatment, in addition to the related psychological, social, and spiritual problems. Palliative care is also called comfort care, supportive care, and symptom management.
Palliative care is provided throughout a patient's experience with cancer. It usually begins at diagnosis and continues through treatment, follow-up care, and the end of life.
External links
- www.bnos.org.uk – British Neuro-Oncology Society (BNOS)
- www.cochrane.org – Trusted evidence. Informed decisions. Better health.
- www.soc-neuro-onc.org – Society for Neuro-Oncology
References
- ^ Levin, VA (April 1999). "Neuro-oncology: an overview". Archives of Neurology. 56 (4): 401–4. doi:10.1001/archneur.56.4.401. PMID 10199326.
- ^ a b c d e f McAllister, L.D., Ward, J.H., Schulman, S.F., DeAngels, L.M. (2002). Practical Neuro-Oncology: A Guide to Patient Care. Woburn, MA: Butterworth-Heinemann.
- ^ Smits, A. (2011). Seizures and the natural history of World Health Organization grade II gliomas: a review. Neurosurgery (2011): 1326-1333.
- ^ Read, Tracy-Ann; Hegedus, Balazs; Wechsler-Reya, Robert; Gutmann, David H. (July 2006). "The neurobiology of neurooncology". Annals of Neurology. 60 (1): 3–11. doi:10.1002/ana.20912. PMID 16802285. S2CID 870084.
- ^ a b c d e f g h i j k l m n o Liu, James K.; Patel, Smruti K.; Podolski, Amanda J.; Jyung, Robert W. (September 2012). "Fascial sling technique for dural reconstruction after translabyrinthine resection of acoustic neuroma: technical note". Neurosurgical Focus. 33 (3): E17. doi:10.3171/2012.6.FOCUS12168. PMID 22937851.
- ^ a b c d Muller, H. L., Gebhardt, U., Warmuth-Metz, M., Pietsch, T., Sorensen, N., & Kortmann, R. D. (2012). Meningioma assecond malignant neoplasm after oncological treatment during childhood. 188, 438-441. Retrieved from [1][dead link]
- ^ a b c d e Ansari, Shaheryar F.; Terry, Colin; Cohen-Gadol, Aaron A. (September 2012). "Surgery for vestibular schwannomas: a systematic review of complications by approach". Neurosurgical Focus. 33 (3): E14. doi:10.3171/2012.6.FOCUS12163. PMID 22937848. S2CID 46630604.
- ^ Cha, Soonmee (July 2009). "Neuroimaging in neuro-oncology". Neurotherapeutics. 6 (3): 465–477. doi:10.1016/j.nurt.2009.05.002. PMC 5084183. PMID 19560737.
- ^ a b Duffau, H. (2012). The challenge to remove diffuse low-grade gliomas while preserving brain functions. 10(7), 569-574.[dead link]
UpToDate Contents
全文を閲覧するには購読必要です。 To read the full text you will need to subscribe.
- 1. 限局性脳幹部神経膠腫 focal brainstem glioma
- 2. What's new in oncology
- 3. 神経学の最新情報 whats new in neurology
English Journal
- Outcome and molecular characteristics of adolescent and young adult patients with newly diagnosed primary glioblastoma: a study of the Society of Austrian Neurooncology (SANO).
- Leibetseder A, Ackerl M, Flechl B, Wöhrer A, Widhalm G, Dieckmann K, Kreinecker SS, Pichler J, Hainfellner J, Preusser M, Marosi C.SourceDepartment of Medicine I, Division of Oncology, Medical University of Vienna (A.L., M.A., B.F., C.S., M.P., C.M.); Institute of Neurology, Medical University of Vienna (A.W., J.H.); Department of Neurosurgery, Medical University of Vienna (G.W.); Department of Radiotherapy and Radiobiology, Medical University of Vienna (K.D.); Landes-Nervenklinik Wagner-Jauregg, Linz, Austria (S.-S.K., J.P.); and Comprehensive Cancer Center - Central Nervous System Tumours Unit (CCC-CNS), Medical University of Vienna, Vienna, Austria (A.W., G.W., K.D., J.H., M.P., C.M.).
- Neuro-oncology.Neuro Oncol.2012 Dec 7. [Epub ahead of print]
- BackgroundYoung age is a favorable prognostic factor for patients with glioblastoma multiforme (GBM). We reviewed the outcomes and molecular tumor characteristics of adolescent and young adult patients with GBM treated in 2 Austrian centers.Patients and MethodsData on patients with histologically pr
- PMID 23223340
- Multimodal MR Imaging (Diffusion, Perfusion, and Spectroscopy): Is It Possible To Distinguish Oligodendroglial Tumor Grade and 1p/19q Codeletion in the Pretherapeutic Diagnosis?
- Fellah S, Caudal D, De Paula AM, Dory-Lautrec P, Figarella-Branger D, Chinot O, Metellus P, Cozzone PJ, Confort-Gouny S, Ghattas B, Callot V, Girard N.SourceCentre de Résonance Magnétique Biologique et Médicale, Center for Research in Oncobiology and Oncopharmacology (CRO2), and Mathematics Department, Aix-Marseille University, Marseille, France; and Departments of Neuroradiology, Pathology and Neuropathology, Neurooncology, and Neurosurgery, APHM, Hopital de la Timone, Marseille, France.
- AJNR. American journal of neuroradiology.AJNR Am J Neuroradiol.2012 Dec 6. [Epub ahead of print]
- BACKGROUND AND PURPOSE:Pretherapeutic determination of tumor grade and genotype in grade II and III oligodendroglial tumors is clinically important but is still challenging. Tumor grade and 1p/19q status are currently the 2 most important factors in therapeutic decision making for patients with thes
- PMID 23221948
Japanese Journal
- 覚醒下手術がもたらしたもの(<特集>悪性グリオーマ治療の進歩)
- 脳神経外科ジャーナル 19(12), 907-915, 2010-12-20
- NAID 110007989508
- MicroRNA-10b regulates tumorigenesis in neurofibromatosis type 1
- Cancer science 101(9), 1997-2004, 2010-09-10
- NAID 10027831452
- 眼球内でかなり進行した網膜芽細胞腫の場合に, どのように対応すべきか
- 眼科臨床紀要 = Folia Japonica de ophthalmologica clinica 1(4), 352-359, 2008-04-15
- NAID 10024295414
Related Links
- Professional society dedicated to promoting advances in neuro-oncology through research and education.
- The Journal of Neuro-Oncology is a multi-disciplinary journal encompassing basic, applied, and clinical investigations in all research areas as they relate to cancer and the central nervous system. It provides a single forum for ...
Related Pictures








★リンクテーブル★
| リンク元 | 「神経腫瘍学」 |